بیماری شکارچی سالمندان؛ فرضیه آمیلوئید در حال آزمایش؛ علل آلزایمر
فرضیه آمیلوئید در حال آزمایش
با پیشرفت درمان های آلزایمر، آیا اکنون زمان آن فرا رسیده است که محققان به لیست علل احتمالی این بیماری موارد بیشتری بیفزایند؟
در اوایل سال 2018، دو آزمایش بالینی دارویی بزرگی که بر روی بیماری آلزایمر انجام می شد، با ناامیدی متوقف گردید. این داروها هم به لیست بلند بالای درمان هایی ملحق شدند که هیچ فایده قابل توجهی برای مبتلایان به این بیماری نداشته اند. فرضیه آمیلوئید که فرض می کند تجمع پپتید آمیلوئید – β عامل اصلی این بیماری می باشد، به مدت بیش از 25 سال در تحقیقات بر روی آلزایمر فرضیه غالب محسوب می شده است. محققان ادعا می کردند که هنگامی که آمیلوئید – β در مغز اجتماع می کند، رسوباتی را به جای می گذارد که منجر به تخریب عصبی و نهایتاً از دست دادن حافظه و توانایی شناختی می گردد. بنابراین آمیلوئید – β به یک هدف درمانی آشکار تبدیل شد – اگر با این پپتیدها مقابله نمایید، خواهید توانست بیماری را نیز درمان کنید.
فرضیه آمیلوئید هرگز در سطح جهانی پذیرفته نشد و آزمایشات ناموفق دارویی تنها انتقادهای منتقدان را به دنبال داشته است. جان هاردی، متخصص مغز و اعصاب دانشگاه کالج لندن و یکی از پیشگامان این فرضیه می گوید: “این فرضیه منجر به ترکیبی از تفکرات محتاطانه و ابهام در مورد این که آیا تحقیقات در مسیر درستی قرار دارند یا خیر شده است”. با این حال نه او نه خیلی دیگر از محققان هنوز امید خودشان را از دست نداده اند. دنیس سلکوئی نورولوژیست دانشکده پزشکی هاروارد در بوستون، ماساچوست می گوید: “همه ما ناامید گشته ایم، اما برای هر کدام از شکست ها دلایل واضحی وجود دارند”. این دلایل می توانند به طور ساده داروهایی باشند که خیلی دیر و پس از پیشرفت کامل بیماری تجویز می شوند و قادر نیستند تا آسیب های به وجود آمده در شرکت کنندگان در آزمایش های ما را جبران نمایند.
همان طور که سلکوئی اشاره کرده است، این حوزه از آزمایشات اکتشافات بسیار ناامید کننده ای به دنبال داشته اند که در ابتدا به عنوان درمان های موثری تلقی می شدند. با این حال حتی اگر ایده های اصلی نیز اشتباه باشند، محققان حداقل به انتهای یک کوچه بن بست نزدیک شده اند. آزمایشات جاری امید زیادی را برای افرادی که در معرض خطر بیماری آلزایمر قرار دارند اما هنوز هیچ علائمی را مشاهده نکرده اند، به وجود آورده اند، علی الخصوص افرادی که نوع ارثی این بیماری را دارا هستند. با این حال آمیلوئید – β تنها علت اصلی بیماری نمی باشد و برخی از محققان فکر می کنند که زمان کشف علت های جدید بیماری فرا رسیده است.
یگ نظریه قدیمی
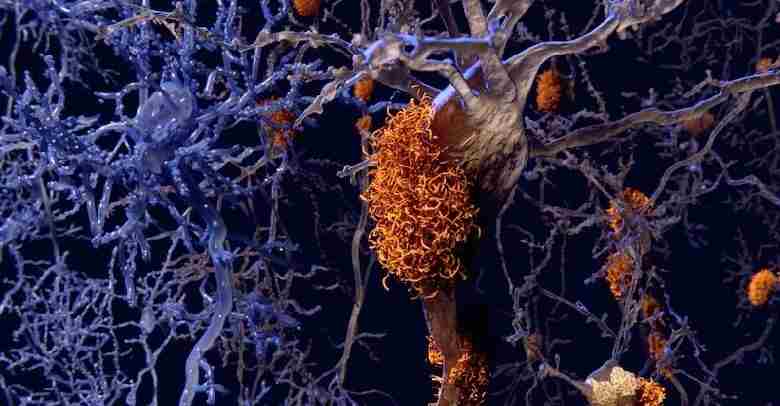
مغز افرادی که به بیماری آلزایمر مبتلا هستند دارای دو مشخصه سلولی می باشد: توده های آمیلوئید – β که معروف به پلاک هستند و در خارج از سلول ها ایجاد می شوند و همچنین رشته های پروتئینی به نام تاو که با عنوان توده های نوروفیبریلاری نیز شناخته می شوند و در درون سلول ها تشکیل می شوند. هر دو این رسوبات برای اولین بار در بیش از یک قرن پیش توضیح داده شدند، تا زمانی که جورج گلنر، پاتولوژیست دانشگاه کالیفرنیا، سن دیه گو، آمیلوئید – β را مورد آزمایشات جداگانه ای قرار داد. گلنر به همراه همکارش کین وونگ نشان داد که این پروتئین از پروتئین بزرگتری به نام پروتئین پیش ساز آمیلوئید (APP) ساخته شده است که در غشای سلولی قرار گرفته و می تواند از آن عبور کند. تحقیقات بعدی آن ها فرآیند تولید آمیلوئید – β را نشان می داد: آنزیم های β – سکرتاز و γ – سکرتاز، پروتئین پیش ساز APP را به دو قسمت تقسیم کرده و یک قطعه پپتیدی به وجود می آورند (یک تقسیم عمیق تر).
نمودار نشان می دهد که چطور آمیلوئید – β از پروتئین پیش ساز آمیلوئید تشکیل می شود.
گلنر تصور می کرد که آمیلوئید – β می تواند عامل بیماری آلزایمر باشد، اما فرضیه آمیلوئید تا زمان کشف جهش های موروثی غالب که اشکال خانوادگی این بیماری را سبب می شوند، کامل نشده بود. یک تیم به رهبری هاردی اولین جهش در ژن APP را در سال 1991 مورد ارزیابی قرار داد. دو مورد دیگر نیز در سال 1996 و در ژن هایی که پروتئین های γ – سکرتاز پریسنیلین 1 (PSEN1) و پریسنیلین 2 (PSEN2) را کدگذاری می کردند شناسایی شدند و مشاهده گردید که ناحیه تقسیم γ – سکرتاز از APP را تحت تاثیر قرار می دهند. این جهش ها منجر به تولید انواع طولانی تری از آمیلوئید – β می شوند که قادر هستند به راحتی در کنار هم اجتماع یابند. متعاقباً این فعل و انفعالات باعث تجمع آمیلوئید – β در ساختارهای بزرگتری به نام الیگومر می شود. تجمل بیشتر آن ها منجر به تولید فیبریل های نامحلول می شود که در نهایت پلاک هایی را تشکیل می دهد که عامل بیماری آلزایمر محسوب می شود. این پلاک ها اغلب توسط هاله ای الیگومرهای محلول احاطه شده اند.
این واقعیت که پلاک های حاوی آمیلوئید – β جهش های ناشی از تجمع آمیلوئید – β را افزایش می دهند باعث شده است که آمیلوئید – β به عنوان عامل اصلی آلزایمر ارثی شناخته شود. میشل گودرت، نورولوژیست دانشگاه کمبریج، انگلستان می گوید: “این دو در کنار همدیگر یک استدلال قوی به وجود می آورند.” تصور می شود که تجمع آمیلوئید – β باعث ایجاد مجموعه ای از فرآیندهای بیماری زا مانند التهاب، تشکیل توده های درهم تنیده تاو، ایجاد اختلال در سیناپس ها و مرگ سلولی می گردد که نهایتاً منجر به زوال عقل می شود. نوع خانوادگی بیماری آلزایمر تقریباً از سایر اشکال این بیماری قابل تشخیص نمی باشد، با این تفاوت که نوع ارثی آن نادر بوده و زودتر از نوع دیگر ایجاد می شود و از این رو این ایده که آمیلوئید – β اولین گام در تشخیص بیماری می باشد، تمام انواع آلزایمر را دربر می گیرد.
آزمایش شواهد
یک استدلال رایج برای فرضیه آمیلوئید این است که پلاک ها در مغز بسیاری از افراد مسن با وضعیت شناختی نرمال نیز یافت می شوند. با این حال طرفداران این ایده یک استدلال پایداری دارند مبنی بر این که: افراد به ظاهر سالمی که این پلاک ها را در مغزشان دارا هستند، ممکن است در مرحله پیش از ظهور علائم بیماری قرار داشته باشند. آن ها همچنین می گوید که همه این پلاک ها مشابه هم نیستند. مطالعات بسیاری انجام شده است که نشان می دهد الیگومرهای محلول سمیت بیشتری نسبت به پلاک های رسوب شده دارند، علی الخصوص به لحاظ آسیب های سیناپسی در مناطقی از مغز که زیربنای حافظه را تشکیل می دهند. یک مطالعه در سال 2013 نشان داد که ارزیابی غلظت الیگومرهایی که به شکل هاله ای دور پلاک ها را احاطه کرده اند، در تمایز نوع زوال عقل بیمارانی که پلاک هایشان متفاوت از هم می باشد، استفاده می شود. با توجه به این امر که الیگومرهایی را که به لحاظ بیولوژیکی فعال تر هستند منجر به ایجاد پلاک می شوند، می توان این گونه استدلال کرد که پلاک ها در ابتدا نقش محافظت از مغز را داشته اند و سپس منجر به بیماری آلزایمر شده اند.
ارتباط میان غلظت پلاک ها با شدت بیماری نیز بر پایه فرضیه اصلی آمیلوئید می باشد. علائم شناختی بیماری بیشتر با تعداد و محل توده های درهم تنیده تاو مرتبط هستند تا با پلاک های آمیلوئید – β. علاوه بر این شواهد حاصل از آزمایشات پس از مرگ نیز نشان می دهند که چنین توده های درهم تنیده ای اغلب در غیاب پلاک ها تشکیل می شوند. این مشاهدات به بحث در مورد رابطه بین این رسوبات با همدیگر منجر شده است، به گونه ای که برخی از محققان ادعا می کنند که آن ها به طور مستقلی در بروز بیماری آلزایمر نقش دارند. آزمایشات بر روی موش ها و خطوط سلولی انسان ها نشان داده است که برخی از آسیب های عصبی مرتبط با این بیماری، مربوط به پروتئین تاو می باشد. با این حال برخی دیگر نیز نشان داده اند که جهش هایی که منجر به تجمع آمیلوئید – β می شود، اجتماع توده ها را تشدید می کنند و عکس آن نیز صادق نمی باشد – جهش هایی که بر روی اجتماع تاو اثر می گذارند، باعث تجمع آمیلوئید – β نمی شوند. بنابراین برخی از محققان ادعا کرده اند که آمیلوئید – β نیروی محرکه اصلی در بیماری آلزایمر می باشد و افزایش میزان تاو نیز علائم بیماری را تشدید کرده و باعث آسیب های سلولی می گردد. گودرت می گوید: “این پایه و اساس فرضیه آمیلوئید می باشد”.
با این حال بزرگترین اعتراضات به این فرضیه، مربوط به آزمایشات بالینی بر روی داروهایی است که آمیلوئید – β را هدف قرار می دهند. رویکردهای درمانی مختلفی در این زمینه مورد ارزیابی قرار گرفته اند مانند ایمونوتراپی و مهار β – سکرتاز یا γ – سکرتاز. اولین آزمایش ایمونوتراپی که به منظور تحریک پاسخ ایمنی بدن طراحی شده بود و مولکول های آمیلوئید – β را هدف قرار می داد، در سال 2002 و پس از این که 6% از شرکت کنندگان دچار التهاب مغز و مننژ شدند متوقف گردید. داروی سماگاستست که تنها مهار کننده γ – سکرتاز بوده است و به مرحله سوم آزمایشات نیز راه یافته است، تاثیرات به مراتب بدتری داشت. آزمایشات در سال 2011 به دلیل افزایش ابتلا به سرطان پوست در میان دریافت کنندگان این دارو متوقف گردید و در عین حال نیز به نظر می رسید که این دارو وضعیت شناختی شرکت کنندگان را بدتر هم کرده است. بیمارانی که از داروی سولانزوماب با عنوان ایمونوتراپی غیر فعال استفاده کرده بودند که آمیلوئید – β را هدف قرار می داد، علیرغم اثربخشی دارو در آزمایشات بالینی، کاهش قابل توجهی نیز در تحلیل قدرت شناختی در مرحله III از خودشان نشان دادند. آزمایشات اخیر فاز III بر روی داروی مهار کننده β – سکرتاز با عنوان وروبکستات (Verubecestat) نیز ناموفق بوده است.
در برخی از موارد داروها به طور کلی بی اثر بودند. داروی سماگاکستات وضعیت دریافت کنندگان این دارو را بدتر کرده بود چرا که علاوه بر APP، γ – سکرتاز، پروتئین های دیگری را نیز تجزیه کرده بود که برخی از آن ها در عملکردهای بیولوژیکی نقش حیاتی ایفا می کنند. از طرفی نیز به نظر نمی رسید که محدود کردن فعالیت آنزیم ها ایده خوبی باشد؛ در مقابل محققان در حال کار بر روی APP هایی بودند که γ – سکرتاز را تقسیم کرده و پپتیدهای کوتاه تری را به وجود می آورد که فعالیت های آنزیم ها را نیز دچار اشکال نمی ساخت. همچنین داروی آدوکانوماب در مرحله III آزمایشات نتایج امیدوار کننده ای نشان داد. آدوکانوماب به پاکسازی کامل آمیلوئید – β از مغز، تنها باعث پاک شدن پلاک ها شده بود. آزمایشات اولیه در سال 2016 نشان داد که این دارو میزان تحلیل قدرت شناختی بیمارانی که در مراحله اولیه بیماری قرار داشتند، را کندتر کرده بود. سلکوئی می گوید: فواید این داروها با دوز تجویز شده ارتباط مستقیمی داشت، که این همان چیزی است که هر دانشمندی می خواهد به آن پی ببرد. نتایج مرحله III از یک کارآزمایی در سال 2020 یافته های فوق را پیش بینی کرده بودند. در صورت موفقیت هر یک از این شیوه های درمانی که آمیلوئید – β را هدف قرار می دهند، راه عبور از این مانع بزرگ باز می شود.
با این حال بزرگ ترین دلیل برای شکست چنین آزمایشاتی این است که این داروها پس از پیشرفت بیماری تجویز می شوند. اسن های توموگرافی گسیل پوزیترون از مغز نشان می دهند که پلاک ها ممکن است دهه ها پیش از بروز علائم شناختی در افراد تشکیل شوند و نیز سطح آمیلوئید – β در هنگام آزمایشات اولیه قابل توجه نباشد. هاردی می گوید: دیدگاه اصلی این است که تمام این داروها، داروهای مناسبی هستند اما برای تجویز و اثربخشی خیلی دیر شده است. به گونه ای که گویا پس از بروز حمله قلبی، برایش استاتین تجویز شود. مایکل مورفی نورولوژیست دانشگاه کنتاکی، لکسینگتون در آمریکا می گوید: اگر فرآیند اجتماع توده های آمیلوئید – β عامل اصلی بیماری باشد، در این صورت ما اگر داروهای از بین برنده آن ها را به بیماران 50 ساله بدهیم، احتمالاً قادر خواهیم بود تا بیماری آلزایمر را درمان کنیم.
چندین طرح برای آزمایش داروهایی که آمیلوئید – β را هدف قرار می دهند بر روی افرادی که در معرض ابتلا به آلزایمر قرار دارند، پیش از بروز علائم بیماری در حال اجرا هستند. شبکه Dominantly Inherited Alzheimer که در دانشکده پزشکی واشنگتن در سنت لوئیس، میسوری واقع می باشد، در حال آزمایش بر روی افرادی است که جهش APP، PSEN1 یا PSEN2 در آن ها تشخیص داده شده است و در مرحله اولیه از بیماری قرار دارند. شرکت کنندگان ابتدا به مدت چهار سال با آنتی بادی هایی نظیر سولانزوماب تحت درمان قرار می گیرند، با این حال محققان همچنان در حال آزمایش سایر داروها نیز هستند. سولانزوماب همچنین توسط یک مطالعه A4 که توسط دانشگاه کالیفرنیای جنوبی در لس آنجلس انجام می شود، مورد ارزیابی قرار گرفته است. این دارو به مدت سه سال برای بیماران 65 تا 85 ساله ای که با توجه به نتایج اسکن های توموگرافی گسیل پوزیترون، به دلیل سطح بالای آمیلوئید – β در مغزشان در معرض ابتلا به زوال عقل قرار دارند، تجویز گردید. عاقبت و آینده فرضیه آمیلوئید بر روی نتیجه این آزمایشات متکی می باشد. مورفی می گوید هنگامی که داروهای از بین برنده آمیلوئید – β به بیماران مبتلا به آلزایمر تجویز می شود و هیچ تاثیر مثبتی مشاهده نمی شود، من به طور کامل متقاعد می شوم که ما در مسیر اشتباهی قرار داریم و گمراه شده ایم.
فرصت هایی که نادیده گرفته شده اند
آمیلوئید – β به مدت دهه های متمادی فرضیه غالب در تحقیقات بر روی بیماری آلزایمر بوده است – احتمالاً با اهمیت بیشتری در مقایسه با فرضیه های ارزشمند دیگر. حتی حامیان سرسخت فرضیه آمیلوئید نیز تصور می کنند که آمیلوئید – β تنها هدفی نیست که باید بر روی آن تمرکز کرد، علی الخصوص پس از این که پیشرفت بیماری پس از حذف تجمعات آمیلوئید – β همچنان ادامه یافته است. با این حال به تحقیق و بررسی سایر داروها نیز توجهی نشده است.
اکثر محققان پیشنهاد می کنند که آمیلوئید – β باعث تسریع تشکیل توده های تاو می شود و تاو نیز بیشتر از آمیلوئید – β به نورون ها آسیب می رساند. به همین دلیل و علیرغم شکست در فاز سوم آزمایشات دارویی در سال 2016 که آمیلوئید – β را هدف قرار داده بودند، پروتئین تاو به عنوان یک عامل مهم دیگر در بیماری تلقی گردید. گودرت می گوید: “هر دو آن ها اهداف بسیار خوبی برای پژوهش هستند”. حتی اگر توده های درهم تنیده تاو مستقل از آمیلوئید – β عمل کنند، با این حال یافتن ارتباط بین این دو نتیجه بسیار ارزشمندی برای تحقیقات در پی خواهد داشت. مورفی می گوید، بدین ترتیب اگر چه پاکسازی آمیلوئید – β ممکن است به تنهایی برای بهبود بیماری کمکی نکند، اما اگر آمیلوئید – β و تاو هر دو باهم پاکسازی شوند ممکن است شاهد اثربخشی بسیار بالایی باشیم. اگر من بخواهم بر روی چیزی شرط بندی کنم، قطعاً بر روی همین موضوع خواهد بود.
التهاب نیز به عنوان یک هدف مهم دیگر در نظر گرفته می شود. در مراحل بعدی بیماری آلزایمر، سلول های محافظت کننده از مغز که میکروگلیا نام دارند، دارای فعالیت بیش از حدی بوده و نورون ها را نابود می کنند. برخی از محققان تصور می کنند که این جزو علائم زوال عقل می باشد، بدین معنی که التهاب می تواند یک هدف ارزشمند برای تحقیقات باشد. رودی تانزی نورولوژیست دانشکده پزشکی هاروارد می گوید، اولین گام این است که بررسی شود بیمار در کدام مرحله از بیماری مراجعه کرده است؟ او آمیلوئید – β را به کبریتی تشبیه کرده است که باعث آتش گرفتن بوته ای در جنگل که همان توده های درهم تنیده تاو هستند می گردد و نهایتاً منجر به آتش سوزی در جنگل یا همان مغز می شود. او می گوید: اگر بیمار مبتلا به زوال عقل کامل را با پاکسازی آمیلوئید – β درمان می کنید، مشابه این است که کبریت را پس از آتش گرفتن جنگل خاموش می کنید.
میروگلیا در بخش های مختلفی از مغز وجود دارد. در صورت هدف قرار دادن آمیلوئید، پروتئین های محرک التهاب به نام سایتوکین ها و همچنین برخی مولکول های ناپایداری که به رادیکال های آزاد مشهور هستند ممکن است دچار استرس اکسیداتیو شده و نهایتاً سلول های عصبی آسیب ببینند. اما در برخی روش های دیگر، مواد زائدی که حاصل آمیلوئید – β هستند، از طریق فرآیندی به نام فاگوسیتوز پاکسازی می شوند. بیان ژن CD33 حالت پیش التهابی را فعال می کند، در حالی که TREM2 آن را غیر فعال می کند. از این رو داروهایی که این ژن ها را هدف قرار می دهند قادر هستند تا فرآیند تشکیل توده ها را کند کرده و از طرفی هم ضایعات آمیلوئید – β را پاکسازی نمایند. تانزی می گوید، اگر توانستید میکروگلیا را از حالت پیش التهابی به حالت حفظ فاگوسیتی تبدیل کنید، هر سه پاتولوژی را هدف قرار داده اید. این یک گلوله جادویی می باشد. او در آزمایشاتش نشان داد که کرومولین سدیم که برای درمان آسم به کار می رود، توانست این تغییرات را در موش ها ایجاد کند. این دارو نیز در حال حاضر در مرحله سوم آزمایشات بر روی بیماری آلزایمر قرار دارند و برخی گروه های دیگر نیز رویکرد مشابهی را دنبال می کنند. تانزی می گوید، تقریباً تمام شرکت های داروسازی که بر روی آلزایمر مطالعه می کنند، برنامه CD33 را در لیست کارهایشان قرار داده اند.
علاوه بر اهداف درمانی که بر پایه فرضیه آمیلوئید قرار دارند، برخی از محققان درصدد کشف ایده های جدیدی هستند که فراتر از این فرضیه اصلی هستند. جورج پری نوروبیولوژیست دانشگاه تگزاس در سن آنتونیو می پرسد، آیا ما قرار است کل این میدان مطالعاتی را برای 15 تا 20 سال دیگر به هم وصل کنیم؟
عملکرد طبیعی آمیلوئید – β هنوز در این زمینه مورد توافق قرار نگرفته است و نقش آن به طور کلی عامل بیماری زایی تلقی می شود. در حالی که برخی دیگر تصور می کنند که این اشتباه بزرگ است. پری می گوید، رویکرد فعلی مبتنی بر این ایده می باشد که آمیلوئید – β عامل تمامی مشکل هاست. اما ایده من کاملاً برعکس است. او ادعا می کند که تجمع آمیلوئید – β و تاو در واقع یک پاسخ محافظتی برای فشارهای متابولیک وابسته به سن در مغز می باشد. به عنوان مثال، استرس اکسیداتیو با افزایش سن بیشتر می شود و این به میتوکندری ها که اندامک های تولید انرژی در سلول ها هستند آسیب می رساند. پری تصور می کند که این امر باعث رسوب آمیلوئید – β می شود، چرا که ثابت شده است که کمبود انرژی هم می تواند میزان بیان APP را افزایش بدهد و متعاقباً منجر به تولید اشکال مختلفی از آمیلوئید – β گردد. او به مطالعاتی اشاره می کند که نشان داده اند آمیلوئید – β می تواند استرس اکسیداتیو در مغز را که منجر به اجتماع پپتیدها می شود کاهش دهد. با این حال افزایش مقدار پپتیدها مشکلات دیگری را نیز در پی دارد. او همچنین متوجه شد که آمیلوئید – β محکم به مس متصل شده و توانایی آن را در کاتالیز کردن کاهش داده و سپس رادیکال های آزادی را تولید می کند که باعث آسیب های اکسیداتیو می شوند. پری می گوید این نشان می دهد که داروهایی که عوامل متصل شونده به مس را مسدود می کنند، استرس اکسیداتیو را کاهش می دهند و سطح آمیلوئید – β را تنظیم می کنند می تواند برای درمان مفید باشند.
برخی از محققان تصور می کنند که عفونت های خاصی می توانند باعث بروز بیماری آلزایمر بشوند که این نشان می دهد داروهای ضد میکروبی یا ضد ویروسی ممکن است برای آلزایمر ارزش درمانی داشته باشند. آن ها برای شواهدی به دست آورده اند مبنی بر این که عفونت ویروس هرپس سیمپلکس (HSV) که جزو خانواده ویروس تبخال انسانی (HHV) می باشد، ریسک ابتلا به بیماری آلزایمر را بالا برده و برخی از ژن های مرتبط با بیماری آلزایمر مانند AOPE عملکرد سیستم ایمنی را تنظیم می کنند. هرچند HSV در مغز افراد مبتلا و بدون بیماری آلزایمر نیز یافت شده است، با این حال مطالعه ای که در سال 1997 توسط روت ایتزاکی نورولوژیست دانشگاه منچستر در بریتانیا منتشر شد نشان داد که ترکیبی از عفونت های HSV و ژن APOE ε4 می تواند به بروز بیماری کمک کند. ایتزاکی می گوید، این بدین معناست که APOE می تواند با تاثیر بر روی آسیب پذیری بیشتر فرد در مقابل میکروارگانیسم ها، ریسک ابتلا به بیماری آلزایمر را نیز بالا ببرد. یک دهه بعد ایتزاکی پس از تلاش برای دستیابی به بودجه بیشتر برای مطالعات بیشتر نشان داد که عفونت HSV در سلول های انسانی یا موش ها باعث افزایش سطح آمیلوئید – β می شود. او می گوید، ما برای انتشار این نتایج با مشکلات بسیاری مواجه شدیم که ما را وارد یک سری نبرد نمود. او و چند نفر از همکارانش در مقاله ای در سال 2016 که در مجله آلزایمر منتشر کردند ادعا نمودند که نقش میکروارگانیسم ها در بیماری آلزایمر نادیده گرفته شده است و درخواست تحقیقات بیشتری برای آزمایش بر روی عوامل ضد میکروبی بودند. این مقاله اظهار داشت که برخی از میکروارگانیسم ها هنگامی که در داخل مغز افراد مسن با سیستم ایمنی مواجه می شوند، به طور مستقیم و غیر مستقیم از طریق فرآیندهای مربوط به آمیلوئید – β به سلول های مغزی آسیب می رسانند.
منتقدان این ایده ادعا می کنند که ارتباط این عفونت ها و علائم فیزیکی بیماری آلزایمر صرفاً به این دلیل می باشد که هر دو در مغز افراد مسن رخ می دهند. آن ها همچنین به برخی شواهد ژنتیکی اشاره کردند که به نقش عامل آمیلوئید – β در بیماری آلزایمر ارثی اشاره می کند. با این حال نقش غیر مستقیم میکروارگانیسم ها در سال 2016 و پس از این که تانزی و همکارانش مطالعه ای را منتشر کردند که نشان می داد آمیلوئید – β از سلول های انسانی و موش ها در برابر عفونت های باکتریایی محافظت می کند، مورد توجه قرار گرفت. آن ها همچنین مغز موش ها را با باکتری سالمونلا تیفیموریوم آلوده کردند و شاهد تشکیل سریع پلاک ها در مغز شدند. تانزی می گوید، شما می توانید پلاک ها را به سادگی و یک شبه از طریق تزریق میکروب به داخل مغز موش های جوان ایجاد نمایید. او ادعا می کند که آمیلوئید – β یک پپتید ضد میکروبی می باشد. پروتئین های این خانواده به میکروارگانیسم ها متصل می شوند و سپس به الیگومرها و فیبریل هایی تبدیل می شوند که شبکه ای را می سازد که قادر است عوامل عفونت را به دام بیندازد – فرآیندی مشابه با تشکیل پلاک های آمیلوئید – β. این ایده با یافته هایی که ایتزاکی در سال 2009 منتشر کرد مطابقت دارد که نشان می دهد DNA ی HSV در پلاک های مغز افراد مبتلا به آلزایمر وجود دارد. تانزی می گوید، اکنون هیچ یافته جدیدی به جز آن چه که ما نشان دادیم آمیلوئید – β چه نقشی در بدن ایفا می کند وجود ندارد – آن به عنوان یک پروتئین ضد میکروبی کلاسیک عمل می کند. حالا وظیفه ما این است که نشان دهیم این اتفاق چطور در مغز انسان رخ می دهد.
در اوایل ماه جاری محققان به رهبری جوئل دادلی، متخصص ژنتیک از دانشکده پزشکی ایکان در مونت سینای نیویورک، مقاله ای را در مجله Neuron منتشر کردند که نشان می داد دو سویه HHV (HHV-6A و HHV-7) در مغز افراد مبتلا به آلزایمر فراوان تر است. آن ها همچنین پیوندهایی را بین این ویروس ها و شبکه ژن ها و پروتئین های مرتبط با پردازش APP پیدا کردند. در همان شماره از مجله Neuron، تانزی و همکارانش نشان دادند که HHV-6 و HSV-1 قادر هستند پلاک های آمیلوئید – β را در موش ها و عصب های انسانی 3D ایجاد کنند که برای محافظت از مغز در برابر عفونت HSV-13 ترشح شده اند. هرچند که هیچ کدام از این مطالعات به طور قطعی نشان نمی دهند که این ویروس ها حتماً باعث بروز بیماری آلزایمر می شوند، با این حال داده های یک پایگاه داده سلامت در تایوان نشان داد که عفونت HSV نه تنها ریسک ابتلا به این بیماری را افزایش می دهد، بلکه افرادی که با داروهای ضد ویروسی تحت درمان قرار گرفته اند نیز ده بار کمتر از سایر افراد احتمال دارد که به بیماری آلزایمر مبتلا شوند.
تانزی تلاش می کند تا ادعای خودش را از سایر ادعاها مبنی بر این که میکروارگانیسم ها مستقیماً مسئول آسیب های ناشی از بیماری آلزایمر هستند متمایز سازد. “ما این را نمی گوییم؛ ما می گوییم که میکروب ها پیش درآمد فرضیه آمیلوئید می باشند. این در واقع یکی از روش های روشن کردن کبریت می باشد”. او اضافه می کند، اشکال ارثی بیماری آلزایمر نتایج دیگری را نشان می دهند، درست مثل نانوذرات موجود در محیط زیست. اگر این ایده تداوم پیدا کند، تانزی می تواند اقدامات پیشگیرانه ای را که از تشکیل پلاک های آمیلوئید – β جلوگیری می کند، پیش بینی نماید.
یک شبکه گسترده
فرضیه آمیلوئید در حال نزدیک شدن به مراتب مهمی می باشد: با آزمایش های متعدد بر روی افرادی که هنوز هیچ علائمی در آن ها دیده نشده است، صحت و یا عدم صحت این یافته ها برای منتقدان آشکار می شود. بسیاری از طرفداران فرضیه آمیلوئید همچنان باور دارند که در مسیر درستی قرار دارند. گودرت می گوید، ما چیزهای زیادی می دانیم. او ادعا می کند که این دانش موجود هنوز فواید بالینی زیادی را ارائه نمی دهد، اما مطمئناً درمان هایی که بر پایه این فرضیه انجام می شوند، اثربخشی خودشان را ثابت خواهند کرد. مورفی می گوید، اگر آزمایش بر روی آدوکانوماب نتیجه مثبتی داشت، ما می توانیم به طرز غیر قابل تصوری به این اهداف نزدیک شویم. اما در صورت عدم موفقیت آن، آزمایشات زیادی لازم خواهد بود تا راه های پیشگیری و درمان بیماری آلزایمر شناسایی شوند. هر موفقیتی در این راه هم برای بیماران و هم برای محققان بسیار جالب توجه خواهد بود؛ گودرت می گوید، حتی فقط یک اثبات برای این اصل می تواند به لحاظ روان شناسی اهمیت زیادی داشته باشد.
اما تا دست یافتن به اولین موفقیت در این زمینه، هنوز چیزهای زیادی برای کشف وجود دارند. مورفی می گوید، ایده های زیادی وجود دارد که باید با جدیت پیگیری و پشتیبانی گردند، در حالی که تعداد کمی از افراد بر روی آن ها کار می کنند. برخی از آن ها ممکن است مهم تر از آنی باشند که ما فکرش را می کنیم. او از اقداماتی نظیر تامین بودجه برای تحقیقاتی که کمتر به آن ها پرداخته شده است حمایت می کند. او می گوید، هیچ چیزی بدتر از این نیست که تنها به فرضیه آمیلوئید پرداخته و اضافه گردد. اکنون زمان آن رسیده است که شبکه های گسترده تری ایجاد کنیم – ما به پایگاهی از ایده های بزرگ تر نیاز داریم.
Nature 559, S4-S7 (2018)
doi: https://doi.org/10.1038/d41586-018-05719-4
This article is part of Nature Outlook: Alzheimer’s disease, an editorially independent supplement produced with the financial support of third parties. About this content.
References
1. Goate, A. et al. Nature 349, 704–706 (1991).
2. Scheuner, D. et al. Nature Med. 2, 864–870 (1996).
3. Haass, C. & Selkoe, D. Nature Rev. Mol. Cell Biol. 8, 101–112 (2007).
4. Esparza, T. J. et al. Ann. Neurol. 73, 104–119 (2013).
5. Nunomura, A. J. et al. Neuropathol. Exp. Neurol. 60, 759–767 (2001).
6. Lövheim, H. et al. Alzheimers Dement. 11, 587–592 (2015).
7. Itzhaki, R. F. et al. J. Alzheimers Dis. 51, 979–984 (2016).
8. Itzhaki, R. F. et al. Lancet 349, 241–244 (1997).
9. Wozniak, M. A., Itzhaki, R. F., Shipley, S. J. & Dobson, C. B. Neurosci. Lett. 429, 95–100 (2007).
10. Kumar, D. K. et al. Sci. Transl. Med. 8, 340ra72 (2016).
11. Wozniak, M. A., Mee, A. P. & Itzhaki, R. F. J. Pathol. 217, 131–138 (2009).
12. Readhead, B. et al. Neuron 99, 64–82 (2018).
13. Eimer, W. A. et al. Neuron 99, 56–63 (2018).
14. Tzeng, N. S. et al. Neurotherapeutics 15, 417–429 (2018).

 ورود/ ثبت نام با جیمیل
ورود/ ثبت نام با جیمیل
