خلاصه
پیشرفت علم به انسان عمر طولانیتری بخشیده است. با این حال، از آنجایی که سلولهای عصبی بازسازی نمیشوند، تعداد افراد مبتلا به اختلالات تخریب عصبی با افزایش سن جمعیت افزایش مییابد. بیماریهای تخریب عصبی در نتیجه از بین رفتن سلولهای عصبی ناشی از عوامل محیطی، جهشهای ژنتیکی، پروتئوپاتیها و سایر اختلالات سلولی رخ میدهند. نقش مستقیم یا غیرمستقیم منفی میکروارگانیسمهای مختلف در شروع یا شدت برخی از اختلالات تخریب عصبی و تعامل بین سیستم ایمنی انسان و میکروارگانیسمهای بیماریزا در این مقاله مروری به تصویر کشیده شده است. این ارتباط ممکن است شروع زودهنگام اختلالات تخریب عصبی را در برخی افراد توضیح دهد. که از طریق مطالعه دقیق پیشینه سلامت این افراد برای ابتلا به هر گونه بیماری میکروبی با میکروارگانیسمهای عصبی بیماریزا (باکتریها، قارچها، ویروسها) قابل ردیابی است. درک و شناخت بهتر رابطه بین میکروارگانیسمها و اختلالات تخریب عصبی ممکن است به محققان در توسعه درمانهای جدید برای اجتناب، به تعویق انداختن یا کاهش شدت اختلالات عصبی کمک کند.
سابقه
تخریب عصبی به از بین رفتن سلول های عصبی، عمدتاً با دلایل ناشناخته، گفته می شود که منجر به اختلالات مختلف سیستم عصبی می شود. بیماری های تخریب عصبی مانند بیماری آلزایمر (AD)، اسکلروز جانبی آمیوتروفیک (ALS)، بیماری هانتینگتون (HD)، مولتیپل اسکلروزیس (MS) و بیماری پارکینسون (PD) به عنوان مسائل بهداشتی مهم در سراسر جهان در نظر گرفته می شوند که به میزان بالای مرگ و میر در افراد کمک می کنند. که با افزایش سن به شدت افزایش می یابد. بر اساس گزارش سازمان جهانی بهداشت (WHO) (Hebert et al., 2013) در بین بیماری های تخریب عصبی، AD و PD به عنوان شایع ترین اختلالات عصبی شناخته شده اند. یک رابطه پیچیده و برجسته بین میکروارگانیسم ها و انسان از میلیون ها سال پیش ایجاد شده است. بسیاری از بیماری های انسانی توسط باکتری های بیماری زا، ویروس ها، قارچ ها و تک یاخته ها ایجاد شده اند. حجم عظیمی از داده ها ثابت کرده اند که عفونت های ناشی از میکروارگانیسم ها در التهاب مزمن در بیماری های تخریب عصبی انسان نقش دارند. میکروارگانیسم ها می توانند از طریق مکانیسم های مختلف باعث اختلال عملکرد CNS و تخریب عصبی شوند. سیستم ایمنی ذاتی میزبان اولین پاسخ بدن در برابر عفونت های میکروارگانیسم هاست که در ساعات اولیه عفونت ایجاد می شود. بر این اساس، فعال شدن سلول های گلیال منجر به تولید مولکول های سیتوکین و کموکاین می شود که با پاسخ التهابی مرتبط هستند (Boulanger, 2009).
میکروبیوتای روده
یک جامعه پیچیده، پویا و بزرگ از میکروارگانیسمها به نام «میکروبیوم» یا «میکروبیوتا» در انسان وجود دارد. تعداد زیادی از این میکروب ها یک مزیت همزیستی یا همزیستی برای میزبان خود دارند. دستگاه گوارش انسان حاوی بزرگترین جامعه میکروارگانیسم ها با حدود ۱۰۱۴ میکروب از ۱۰۰۰ گونه مختلف میکروبی است که دارای حدود ۱۰۶ × ۴ ژن هستند. بیش از ۹۹ درصد از میکروارگانیسم های دستگاه گوارش به عنوان باکتری های بی هوازی با میکروب های دیگر از جمله قارچ ها، تک یاخته ها، ویروس ها و باستانی ها شناسایی شده اند. این جامعه باکتریایی شامل بزرگترین میکروارگانیسم ها با تراکم ۱۰۱۲ در هر میلی لیتر است که بالاترین تراکم میکروبی در هر اکوسیستمی است (Bhattacharjee and Lukiw, 2013). شایع ترین میکروارگانیسم ها در دستگاه گوارش Firmicutes (~۵۱٪) و پس از Bacteroidetes (~۴۸٪) هستند. سایر باکتری ها شامل سیانوباکتریها، فوزوباکتریها، پروتئوباکتریها، اسپیروکتها و وروکومیکروبیا هستند. اخیراً اشاره شده است که میکروبیوتا می تواند بر سلامت و بیماری های انسان تأثیر بگذارد. توسعه روشهای جدید مانند نسلهای جدید فناوریهای توالییابی و بیوانفورماتیک، فرصتی را برای بررسی تعاملات پیچیده میکروارگانیسمها و انسان فراهم کرده است.
ارتباط میکروبیوتای روده و مغز و همچنین نقش کلیدی این ارتباط در رشد مغز و حفظ هموستاز توسط فیزیولوژی مدرن نشان داده شده است. دومی ممکن است از طریق تنظیم پایین ژن های مرتبط با سیستم ایمنی و همچنین تنظیم یکپارچگی سد خونی مغزی به دست آید (Stilling et al., 2015). تعداد فزاینده ای از مطالعات بر نقش مهم میکروبیوتا در تنظیم محور روده-مغز، تعدیل عملکرد مغز و ایجاد مسیری برای ایجاد ارتباط دو طرفه بین مغز و روده تاکید کرده اند (Foster et al., 2016). مطالعات اخیر گزارش داده اند که میکروبیوتای روده از طریق تنظیم غلظت پیش سازها یا تولید انتقال دهنده های عصبی، نقش کلیدی در تنظیم سطوح انتقال دهنده های عصبی دارد. به عنوان مثال، گونه های بیفیدوباکتریوم و لاکتوباسیلوس، انتقال دهنده عصبی مهارکننده گاما آمینوبوتیریک اسید (GABA) را آزاد می کنند. علاوه بر این، تولید نوراپی نفرین توسط گونه های باسیلوس، اشرشیا و ساکارومایسس و سروتونین توسط کاندیدا، انتروکوک و استرپتوکوک گزارش شده است (Lyte, ۲۰۱۴). میکروبیوتای روده متابولیت های مهم دیگری مانند اسیدهای چرب با زنجیره کوتاه (SCFA) را تولید و آزاد می کند. SCFA ها شامل استات، بوتیرات و پروپیونات مهم ترین متابولیت هایی هستند که بر CNS تأثیر می گذارند. واسطه های اعمال SCFAها گیرنده های جفت شده با پروتئین G یا هیستون داستیلازها هستند (Stilling et al., ۲۰۱۶).
مطالعات in vivo نشان دادهاند که موشهای بدون میکروب دارای تنظیم دگرگونی برخی از ژنهای مربوط به پلاستیسیته و مسیرهای متابولیکی خاص از جمله متابولیسم هورمون استروئیدی، تقویت طولانیمدت سیناپسی و سیگنالدهی چرخهای با واسطه آدنوزین ۵ فسفات هستند که بهویژه بر نواحی مخچه و هیپوکامپ تأثیر میگذارد. علاوه بر این، موشهای بدون میکروب افزایش قابلتوجهی در سطوح ۵-HT در هیپوکامپ نشان دادند (Diaz Heijtz et al., 2011; Clarke et al., 2013) به طور جالب توجه، غلظت فاکتور نوروتروفیک مشتق از مغز (BDNF)، پروتئین قابل توجهی در یادگیری، رشد عصبی، حافظه و تنظیم خلق و خوی است. در موش های بدون میکروب کاهش می یابد. شواهد نشان داد که بیان ژن Bdnfدر قشر و آمیگدال موش های بدون میکروب کاهش یافت (Neufeld et al., 2011 ; Clarke et al., 2013). مطالعات بیشتر همچنین نشان داده اند که نوروژنز را می توان توسط میکروبیوتای روده کنترل کرد. به عنوان مثال، نوروژنز هیپوکامپ در موشهای بدون میکروب به دلیل تغییرات در تیتر سوبستراهای مؤثر بر نوروژنز هیپوکامپ، از جمله BDNF، کورتیکوسترون، سیتوکینهای پیش التهابی و سروتونین تحت تأثیر قرار میگیرد (Ogbonnaya et al., 2015). برخی از میکروبیوت های روده مانند گونه های باسیلوس، اشریشیا و ساکارومایسس می توانند نوراپی نفرین تولید کنند. علاوه بر این، سروتونین را می توان توسط کاندیدا، انتروکوک و استرپتوکوک سنتز کرد (Lyte، ۲۰۱۴).
میلیناسیون قشر پیش پیشانی نیز تحت تنظیم میکروبیوتا است. اعتقاد بر این است که موشهای بدون میکروب افزایش حجم آمیگدال را نشان دادند و همچنین هیپرتروفی دندریتیک را در آمیگدال قاعدهای جانبی خود (BLA) نشان دادند. علاوه بر این، تغییرات مورفولوژیکی در شکل نورون های هرمی BLA در موش های بدون میکروب مشاهده شده است (Luczynski et al., ۲۰۱۶). این مشاهدات بر اهمیت کلونیزاسیون میکروبی قبل از شیر گرفتن در روده تاکید می کند.
به طور کلی، میکروبیوتای روده ممکن است به عنوان یک عامل مهم در توسعه CNS و عملکرد بعدی آن شناخته شود. این میکروارگانیسمها برای رفتارهای اضطرابمانند، شناخت، پاسخدهی طبیعی به استرس، اجتماعی بودن و حفظ هموستاز CNS حیاتی هستند. هموستاز CNS مشتق از میکروبیوتا از طریق تنظیم یکپارچگی سد خونی مغزی و عملکرد ایمنی حاصل می شود. نوروژنز، سمیت عصبی، سطح انتقال دهنده عصبی، گیرنده های انتقال دهنده عصبی، سیگنال دهی نوروتروپیک و سیستم های سیناپسی می توانند تحت تأثیر میکروبیوتای روده قرار گیرند.
نقش محافظتی یا منفی میکروبیوتای روده در تخریب عصبی و فرآیندهای رشد عصبی در برخی از بیماری های CNS (جدول ۱) در بخش های بعدی به تفصیل مورد بحث قرار گرفته است.
جدول ۱
اثرات مثبت و منفی میکروبیوتای روده بر عملکرد سیستم عصبی مرکزی (CNS).
The positive and negative effects of gut microbiota on central nervous system (CNS) function.
| Biosynthesized metabolite | Impact on CNS | Function | Type | Gut microbiota example | Disorder |
|---|---|---|---|---|---|
| GABA | Inhibitory neurotransmitter | Inhibitory neurotransmitter reducing neuronal excitability throughout the nervous system | (−) | BifidobacteriumLactobacillus | AD, depression and synaptogenesis impairments |
| BMAA | Neurotoxicity | N-methyl-D-aspartate signaling dysfunction | (−) | Cyanobacteria | AD |
| Oxytocin | Positive-regulation of the neurotransmitters level | Improve social behavior and communication | (+) | Lactobacillus reuteri | Autism |
| – | – | Induction or suppression of autoimmune encephalomyelitis | Both | Different compositions | MS |
| Butyrate | Suppression of inflammation | Suppression of inflammation | (+) | Faecalibacterium Coprococcus | MS |
| Norepinephrine | Positive-regulation of the neurotrasmitters level | Improvement of alertness and arousal, and speeds reaction time | (+) | Bacillus, Escherichia and Saccharomyces species | Depression and hyperactivity disorder |
| Serotonin | Positive-regulation of the neurotrasmitters level | Regulation of anxiety, happiness and mood | (+) | Candida, Enterococcus and Streptococcus | Depression |
| SCFAs | Regulation of synaptic system | Induction of αSynpathology | (+) | – | NDs |
| PUFAs | Appropriate growth and function of nervous tissue | – | (+) | – | Autism |
| – | Reducing the removal of αSyn aggregates | Controlling cellular processes such as autophagy | (−) | – | Lewy body disease, multiple system atrophy and PD |
| Amyloids and LPS | Inducing inflammation |
| (−) | Escherichia coli | AD |
| – | Adverse effects on growth and function of nervous tissue |
|
نقش میکروبیوتای روده در بیماری پارکینسون
آمیلوئیدوز به تجمع غیرطبیعی پروتئین ها در سلول های عصبی گفته می شود و منجر به اختلال در عملکرد سلولی می شود. فرض بر این است که تجمع پروتئین های نامحلول با ترکیب تغییر یافته در بافت ها باعث ایجاد نزدیک به ۵۰ بیماری در انسان می شود. آمیلوئیدوز در برخی از بیماری های تخریب عصبی مانند AD، HD و PD نقش دارد. اعتقاد بر این است که تجمع و تجمع پروتئین α-سینوکلئین (αSyn) در نورونها، بهویژه نورونهای دوپامینرژیک، در سینوکلینوپاتیها از جمله آتروفی سیستم متعدد، بیماری بدن لوی و PD نقش دارد (Brettschneider et al., 2015; Prusiner et al., 2015). بر اساس فرضیه براک (Del Tredici and Braak, 2008)، تجمع پروتئین αSyn در اوایل سیستم عصبی روده (ENS) ظاهر می شود. سپس از طریق عصب واگ به سلول های مغزی منتقل می شود. شواهد پاتوفیزیولوژیک نشان داده است که انتقال αSyn به روده جوندگان سالم می تواند پاتوژنز PD را القا کند. (Sampson et al., 2016) نقش باکتری های روده در پاتوفیزیولوژی سینوکلئینوپاتی ها و ارتباط آن با PD را بررسی کرده اند. نتایج نشان داد که میکروبیوتای روده برای القای نقصهای سنپاتولوژی و حرکتی در مدل موش مورد نیاز است. جالب توجه است که میکروارگانیسم های مدفوعی جدا شده از بیماران مبتلا به PD در مقایسه با افراد سالم اثرات مخرب بیشتری بر عملکرد حرکتی نشان دادند. در موشهای بدون میکروب و تحت درمان با آنتیبیوتیک، تجمع αSyn، نقصهای حرکتی و فعالسازی میکروگلیا در مقایسه با مدلهای حیوانی حاوی میکروبیوتای پیچیده کاهش یافت. یک مکانیسم بالقوه برای آسیب شناسی ناشی از αSyn شامل بلوغ و فعال شدن مسیرهای التهابی میکروگلیا از طریق SCFAs تولید شده توسط باکتری های روده است (Kannarkat et al., 2013). علاوه بر این، التهاب باعث تجمع αSyn، فعال شدن میکروگلیا و پیشرفت اختلالات تخریب عصبی می شود. نشان داده شده است که باکتری روده بر سایر فرآیندهای سلولی مانند اتوفاژی، یک فرآیند ژنتیکی مرتبط با PD، که ممکن است پاکسازی دانه های αSyn را کاهش دهد، تأثیر می گذارد (Beilina and Cookson, 2016).
نقش میکروبیوتای روده در آلزایمر
به رسمیت شناختن اهمیت میکروبیوتای روده در انتشار بتا آمیلوئید (Aβ) در بیماران مبتلا به AD وجود دارد. میکروارگانیسم ها معمولاً برخی از محصولات پیچیده مانند آمیلوئیدها و LPS را دفع می کنند که برای میزبان خود ایمنی زا هستند. در میان آنها، LPS میکروبی می تواند هموستاز میکروبیوتای روده را تغییر دهد و پاسخ التهابی را القا کند، همانطور که در بیماری التهابی روده اتفاق می افتد. علاوه بر این، اعتقاد بر این است که آمیلوئیدهای میکروبی در تجمع، تشکیل بیوفیلم ها، تهاجم و کلون سازی میکروارگانیسمهای بیماریزا نقش دارند. به عنوان مثال، مطالعات آزمایشگاهی به این نتیجه رسیده اند که اشرشیاکلی قادر به ترشح یک اندوتوکسین است که در تشکیل فیبریل های Aβ نقش دارد (Asti and Gioglio, 2014). به طور معمول، آمیلوئیدها و LPS میکروبی یک شکل مونومر و محلول دارند. با این حال، به دنبال تجمع و تشکیل ساختارهای نامحلول، ممکن است با استرس اکسیداتیو و پاتوژنز AD همراه باشند. نقش میکروبیوتا در تشکیل آمیلوئید با افزایش نفوذپذیری اپیتلیوم روده و سد خونی مغزی در طول پیری بیشتر قابل توجه است. همچنین سن با تعداد فیرمیکوت ها و بیفیدوباکتری ها رابطه مستقیم دارد. با این وجود، ترکیب میکروبی روده نیز تحت تأثیر رژیم غذایی، اجتماع و دریافت درمانهای مختلف قرار میگیرد. مطالعات اخیر این فرضیه را مطرح کرده اند که آمیلوئیدهای تولید شده توسط باکتری های روده می توانند از مجرای روده عبور کرده و در مغز تجمع کنند (Zhao and Lukiw, 2015). این امر استرس اکسیداتیو ناشی از آمیلوئیدها را افزایش میدهد و سیگنالدهی فاکتور jB هستهای را فعال میکند که منجر به تنظیم مثبت میکروRNA-34a پیشالتهابی و اختلال فاگوسیتوز پپتید Aβ۴۲ توسط سلولهای میکروگلیال میشود. علاوه بر این، LPS های میکروبی و آمیلوئیدها نشت دستگاه گوارش را تشدید می کنند و باعث تولید سایتوکین های پیش التهابی می شوند که منجر به افزایش شدت AD می شود.
همانطور که قبلا ذکر شد، برخی از باکتری های روده مانند گونه های بیفیدوباکتریوم و لاکتوباسیلوس یک انتقال دهنده عصبی مهاری، یعنی GABA را سنتز و آزاد می کنند. تغییر در سیگنال دهی GABA با افسردگی، AD و اختلالات سیناپتوژنز همراه است. یکی از ساکنان مهم دستگاه گوارش انسان سیانوباکتریهایی است که نوروتوکسینی به نام β -N-methylamino-L-alanine (BMAA) تولید میکنند. BMAA با گیرنده گلوتامات N-متیل-D-آسپارتات تداخل می کند و در نهایت منجر به اختلال در سیگنال دهی N-متیل-D-آسپارتات در بیماری AD و سایر بیماری های تخریب عصبی می شود (Aziz et al., 2013).
نقش میکروبیوتای روده در اوتیسم
اوتیسم یک اختلال عصبی است که با نقص در رفتار و ارتباطات اجتماعی همراه است. اساساً ژنتیک نقش کلیدی در پاتوژنز اوتیسم دارد. با این حال، برخی از اسناد نشان می دهد که بیش از ۷۰ درصد از افراد مبتلا به اوتیسم از سندرم GI رنج می برند که نقش میکروبیوتای روده را در این بیماری نشان می دهد. مطالعات in vivo نشان داده است که موشهای بدون میکروب در مقایسه با گروه کنترل رفتار اجتماعی غیرطبیعی دارند. اگرچه بهبود رفتاری پس از کلونیزاسیون میکروبی مشاهده شده است، اما این رفتار به طور کامل ترمیم نشد (Desbonnet et al., 2015). این ممکن است به دلیل توانایی میکروبیوتای روده در تخمیر SCFA ها باشد که برای تولید اسیدهای چرب غیراشباع چندگانه (PUFA) ضروری هستند. PUFA ها برای رشد مغز از نظر رشد و عملکرد مناسب بافت عصبی مهم هستند. سطوح پایین PUFA ممکن است با اختلالات رشد عصبی، به عنوان مثال اوتیسم، و با مشکل در عملکرد رفتاری و شناختی مرتبط باشد (El-Ansary and Al-Ayadhi, 2014).
به طور جالب توجهی، بررسی میکروبیوتای مدفوع، تغییراتی را در جامعه میکروبی روده در بیماران اوتیسم نشان داده است. بر اساس تجزیه و تحلیل تنوع میکروبی، نشان داده شده است که نسبت Bacteroidetes به Firmicutes کاهش می یابد در حالی که جمعیت گونه های Lactobacillus و Desulfovibrio افزایش می یابد (Tomova et al., ۲۰۱۵).
برخی از میکروبیوت های روده مانند باکتری پروبیوتیک Lactobacillus reuteri می توانند سطح اکسی توسین را به میزان قابل توجهی افزایش دهند (Erdman and Poutahidis, ۲۰۱۴). اکسی توسین یک پپتید ضروری است که توسط هیپوتالاموس ترشح می شود و در رفتار و ارتباطات اجتماعی نقش دارد. حذف ژن گیرنده اکسی توسین منجر به کمبود رفتاری قابل توجهی در مدلهای موش شد که نقش کلیدی میکروبیوتای روده را در بهبود رفتار ثابت کرد.
نقش میکروبیوتای روده در ام. اس.
مطالعات اخیر ترکیب میکروبیوتای روده را با شدت MS مرتبط کرده است. مطالعات in vivo در مدل موش ام اس توانایی باکتری های روده را در القای آنسفالومیلیت خودایمنی پس از قرار گرفتن در معرض گلیکوپروتئین الیگودندروسیت میلین نشان داده است (Berer et al., ۲۰۱۱). در مطالعه دیگری (Cantarel et al., 2015)، جامعه باکتریایی روده بیماران ام اس پس از درمان با ویتامین D با افراد سالم مقایسه شد. افراد نسبت به افراد سالم پس از درمان بیماران با ویتامین D، جمعیت آکرمانسیا، کوپروکوکوس وگونه های فکالی باکتریوم در GI افزایش یافت. این یافته مطابق با این واقعیت است که Faecalibacterium به دلیل توانایی خود در تولید بوتیرات می تواند در سرکوب التهاب نقش داشته باشد. کوپروکوکوس نیز مانند فاکالی باکتریوم ممکن است به عنوان یک باکتری کاهش دهنده التهاب در نظر گرفته شود (Cantarel et al., 2015). بررسیهای جامعه میکروبیومی بیماران اماس عودکننده- فروکشکننده و افراد سالم نیز توسط چن و همکاران انجام شده است. (Chen et al., 2016). در افراد قبلی، جمعیت جنسهای Blautia، Dorea، Haemophilus، Mycoplana و Pseudomonas افزایش یافت، در حالی که در افراد دوم، افزایش جمعیت Adlercreutzia، Parabacteroides و Prevotella مشاهده شده است. به طور خاص، غنای گونه ای در افراد سالم در مقایسه با بیماران ام اس افزایش یافته است. این مطالعات بر دیس بیوز میکروبی روده در بیماران ام اس و در نتیجه اهمیت میکروبیوتای روده در سبب شناسی و پاتوژنز ام اس تاکید می کند.
باکتریها
مایکوباکتریوم لپره
مایکوباکتریوم لپره یک پاتوژن است که با هدف قرار دادن و دستکاری ساختار و عملکرد سلول های شوان در عصب محیطی، مسئول دمیلینه شدن و آسیب عصبی است. M. leprae در ابتدا می تواند به یک گلیکوپروتئین ۲۸ کیلو دالتون، میلین P صفر (P0)، یک پروتئین اصلی عصب محیطی انسان که به طور خاص در آنها بیان می شود، متصل شود (Vardhini et al., ۲۰۰۴). اتصال مولکولهای ساختاری مشابه M. leprae میتواند برهمکنشهای P0-P0 منجر به دمیلینه شدن، فرآیندی که معمولاً به عنوان دمیلینهسازی وابسته به تماس نامیده میشود، مختل کند. نوروپاتولوژی M. lepraeبا تغییر در محیط سلول های شوان و فعال شدن مسیرهای آپوپتوز در سلول ها، ویژگی جذام، آغاز می شود. این فرآیند پاسخ خودایمنی میزبان را در برابر آنتی ژن های سلول های عصبی و به دنبال آن دمیلیناسیون و مرگ سلولی ایجاد می کند (شکل ۱). مطالعات بیوانفورماتیک شباهت بین پروتئین M. leprae و عصب محیطی انسان، یعنی محل اتصال این باکتری را آشکار کرده است (Vardhini et al., ۲۰۰۴). به طور خاص، یک توالی ۱۷-۴۲-اسید آمینه در پروتئین خانواده P60 ترشح شده M. leprae شباهت به دنباله اسید آمینه ۵۱-۵۶ در میلین P0 (در دامنه ایمونوگلوبولین) دارد. این شباهت نقش P0 را در تعاملات پروتئین-پروتئین و پروتئین-لیگاند و همچنین عوارض خودایمنی در سیستم عصبی نشان می دهد (Zhu et al., 2001). شایان ذکر است که دامنه ایمونوگلوبولین از طریق خاصیت چسبندگی هموفیلیک نقش مهمی در برهمکنش بین پروتئین ها و لیگاندها ایفا می کند (Brümmendorf and Lemmon, 2001).
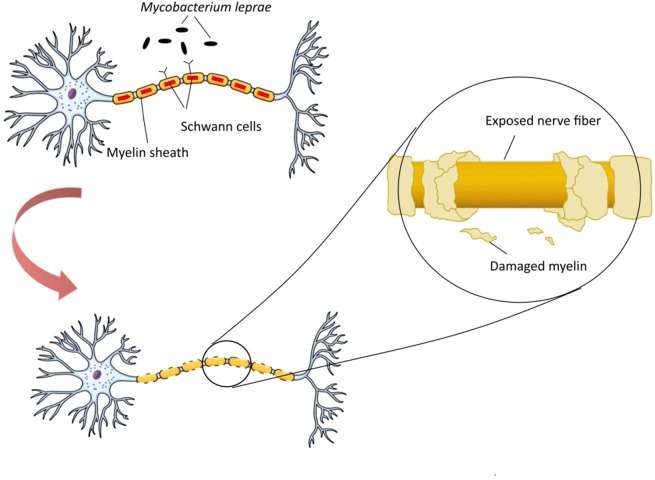
گونههای مایکوپلاسما
گونه های مایکوپلاسما عامل بیماری های تنفسی در انسان هستند. در حال حاضر نقش این باکتری در اختلالات CNS مورد توجه قابل توجهی قرار گرفته است. مایکوپلاسما را می توان با استفاده از کشت مستقیم، PCR و وسترن بلات در نمونه ها تشخیص داد. دو روش آخر آنتیبادیهای IgM و IgG را در برابر پروتئینهای غشایی مرتبط با لیپید (LAMP) شناسایی میکنند. وجود مایکوپلاسما در جریان خون بیماران ALS توسط گیل و همکاران بررسی شده است. (Gil et al., 2014). بر اساس این مطالعه، گونه های مایکوپلاسما در ۴۶ درصد از بیماران ALS و ۹ درصد افراد سالم با استفاده از روش های کشت یا PCR مستقیم شناسایی شد. آنها همچنین سرم را برای تشخیص IgM- و IgG-ویژه لامپ های مایکوپلاسما فرمنتانس بررسی کردند.. ۴۶ و ۳۱ درصد از بیماران مبتلا به ALS به ترتیب IgM و IgG را در برابر LAMP های M. fermentans نشان دادند ، در مقایسه با ۷ درصد برای هر یک از آنتی بادی ها در افراد سالم. برخی از نمونه های خون بیمار (۴۶%) و افراد سالم (۹%) نیز آلودگی به مایکوپلاسما را نشان دادند. به طور جالبی، M. fermentans مایکوپلاسما SP شناسایی شده بود. در تمام بیماران مثبت جنس مایکوپلاسما و در ۵۰ درصد افراد سالم مثبت برای مایکوپلاسما (Gil et al., 2014). به طور مشابه،(Nicolson et al., 2002) نمونه های خون جانبازان جنگ خلیج فارس و غیرنظامیان مبتلا به ALS را برای وجود مایکوپلاسما بررسی کردند.گونه ها. در گروه اول، همه افراد دارای عفونت مایکوپلاسمی خون با M. fermentans بودند، به جز یک مورد با M. genitalium . در مقابل، حدود ۷۹٪ (۵۹٪ با M. fermentans) از افراد در گروه دوم حداقل یک Mycoplasma sp.، یعنی M. fermentans ، M. hominis، M. penetrans و/یا M. pneumoniae مثبت بودند. (Nicolson et al., 2002).
به طور کلی، مایکوپلاسما می تواند باعث فعال شدن ماکروفاژها، مونوسیت ها و سلول های گلیال شود که منجر به تولید سیتوکین های التهابی می شود. آنتی ژن های اصلی مایکوپلاسمی LAMP ها هستند که یکی از اهداف مهم پاسخ ایمنی هومورال هستند. مایکوپلاسما ممکن است در پیشرفت ALS و/یا پاتوژنز آن کمک کند. روش دیگر، بیماران مبتلا به ALS ممکن است به شدت در برابر عفونت های سیستمیک با مایکوپلاسما آسیب پذیر باشند .
کلامیدیا پنومونیه
کلامیدیا پنومونیه یک باکتری داخل سلولی است که به طور معمول از طریق مخاط دستگاه تنفسی وارد بدن می شود. این بیماری باعث عفونت های مجاری تنفسی، یعنی برونشیت حاد، برونشیت مزمن و آسم و پنومونی اکتسابی از جامعه در انسان می شود. عفونت C. pneumoniae همچنین در اختلالات CNS مانند AD، مننژوانسفالیت و MS گزارش شده است (Wunderink and Waterer, ۲۰۱۴). دسترسی میکروارگانیسم به CNS از طریق مسیرهای داخل عروقی و بویایی است. حضور این پاتوژن در مایع مغزی نخاعی (CSF) بیماران مبتلا به ام اس پیشرونده یا ام اس عودکننده- فروکش کننده تازه تشخیص داده شده با استفاده از کشت مستقیم، PCR و تشخیص آنتی بادی برای C تایید شد. پنومونیهآنتی ژن های بدن ابتدایی (EB). تقریباً ۸۶ درصد از بیماران MS آزمایش شده سطوح آنتی بادی های آنتی ژن های C. pneumoniae EB را افزایش داده بودند (Sriram et al., ۱۹۹۹).
آنتی ژن های C. پنومونیا همچنین در نئوکورتکس AD و/یا در ارتباط با پلاک های پیری و درهم تنیدگی های نوروفیبریلاری وجود داشتند (Choroszy-Król et al., 2014). در یک مطالعه جداگانه (Balin et al., 1998)، اسیدهای نوکلئیک از نمونههای مغزی پس از مرگ بیماران مبتلا به AD دیررس تهیه و با گروه کنترل مقایسه شد. آزمایشات PCR برای DNA کروموزومی C. pneumoniae برای حدود ۹۰٪ از بیماران AD مثبت بود در حالی که تنها ۵٪ از بیماران کنترل PCR مثبت بودند. علاوه بر این، C. پنومونی می تواند به طور متراکم از برخی از مغزهای AD کشت کند در حالی که همان مطالعات کشت مغز غیر AD برای C. پنومونی منفی بود.. نواحی مغز شامل هیپوکامپ، قشر جداری، قشر جلوی پیشانی و مخچه نیز با استفاده از تکنیکهای میکروسکوپی الکترونی و ایمونوالکترونی مورد بررسی قرار گرفتند. جالب توجه است که اجسام ابتدایی و مشبک کلامیدیا فقط از نواحی مغز AD شناسایی شدند و نه از مغزهای غیر AD (Balin et al., 1998). وجود میکروارگانیسم در مغز نیز توسط ایمونوهیستوشیمی تایید شد. آنتی ژن های C. pneumoniae در آستروسیت ها، ماکروفاژها و میکروگلیاهای هیپوکامپ، قشر جداری و پری فرونتال و قشر گیجگاهی فقط در بیماران مبتلا به AD در مقایسه با گروه کنترل شناسایی شد (Balin et al., ۱۹۹۸ ). در یک مطالعه اخیر (Paradowski et al., ۲۰۰۷)، وجود C. pneumoniae در CSF 57 بیمار AD و ارتباط آن با سطوح A β ۴۲ و پروتئین tau مورد بررسی قرار گرفت و با ۴۷ گروه کنترل مقایسه شد. مشخص شد که حضور DNA کلامیدیا در CSF بیماران مبتلا به AD به طور معنیداری بیشتر از گروه کنترل بود. با این حال، هیچ تاثیری بر سطح پروتئین A β ۴۲ و تاو در CSF نمی تواند با فعالیت میکروارگانیسم مرتبط باشد (Paradowski et al., ۲۰۰۷). این مطالعات به وضوح وجود و زنده ماندن C. pneumoniae را در مغز بیماران مبتلا به AD نشان داد که ممکن است یک عامل خطر برای شروع و/یا پاتوژنز AD باشد.(Gérard et al., 2005) بار C. pneumoniae را مورد مطالعه قرار دادنددر مغز AD بیماران با توجه به آلل نوع ۴ ژنوتیپ آپولیپوپروتئین E (APOE-ɛ۴). آنها بار کمتری از سلولهای آلوده به C. pneumoniae را در نمونههای مغزی بیماران مبتلا به AD که فاقد APOE-ɛ۴ بودند، در مقایسه با مناطق AD-مغز تحت تأثیر آن آلل پیدا کردند. می توان نتیجه گرفت که خطر ابتلا به AD و پیشرفت آن به سمت اختلال عملکرد شناختی در افراد فاقد آلل ɛ۴ کمتر از افراد دارای این آلل است.
نقش منفی C. pneumoniae ممکن است به تولید سیتوکین های پیش التهابی و تجمع Aβ در مغز در طول عفونت های کلامیدیا به دلیل التهاب مزمن و فعال شدن ماکروفاژها و مونوسیت ها نسبت داده شود (شکل .۲A). همانطور که قبلا ذکر شد، مکانیسم اصلی آسیب شناسی AD از تجمع Aβ است. چندین مطالعه گزارش کرده اند که C. pneumoniae اساساً آستروگلیا، ماکروفاژها و میکروگلیا را در بیماران مبتلا به AD که سلول های اصلی مسئول التهاب در مغز هستند، آلوده می کند. التهاب ناشی از C. pneumoniae نقش مهمی در التهاب عصبی درگیر در AD دارد.
 شکل ۲
شکل ۲
ارتباط بین عفونت C. pneumoniae و PD به ندرت مورد مطالعه قرار گرفته است.۰(Turkel et al., 2015) نشان داده اند که ۹۸ درصد از بیماران PD برای C. pneumoniae IgG مثبت بودند، در حالی که C. pneumoniae IgM هم در افراد مبتلا به PD و هم در افراد کنترل منفی بود.
اسپیروکتها
برخی گزارشها نشان دادهاند که اسپیروکتها تأثیر منفی بر پاتوژنز AD دارند. داده های تاریخی نشان می دهد که علائم بیماری AD مشابه علائم پاتولوژیک فلج عمومی است. در یکی از قدیمی ترین مطالعات، نوگوچی و مور (Noguchi & Moore; 1913) امکان وجود ترپونما پالیدوم را در قشر مغز بیماران مبتلا به فلج مورد بحث قرار دادند. آنها نشان داده اند که T. pallidumمی تواند به ایجاد زوال عقل، آتروفی قشر مغز و آمیلوئیدوز در شکل آتروفیک فلج عمومی کمک کند. علائم پاتولوژیک این شکل از پارزی شامل میکروگلیوز، آستروسیتوز و از دست دادن سلول های عصبی است. علاوه بر این، وجود تودههای پلاک مانند کلنیهای باکتریایی در ناحیه قشر مغز شواهد پاتولوژیک مهمی از درگیری T. pallidum در فلج بود. علاوه بر این، تجمع پلاک های آمیلوئید در مغز بیماران مبتلا به فلج عمومی ثبت شده است.
Borrelia burgdorferi یک اسپیروکت منتقل شده از طریق کنه است که باعث بیماری لایم می شود، همچنین در برخی از مغزهای AD شناسایی شد. اولین بروز B. burgdorferi در مغز موارد AD توسط (MacDonald & Miranda; 1987) گزارش شد و شناسایی با استفاده از ویژگی های مورفولوژیکی و ایمونوهیستوشیمی و همچنین روش های سرولوژیکی تایید شد. روشهای سرولوژیکی آنتیبادیهای اختصاصی را در خون، CSF و رگههای نوروفیبریلاری بیماران مبتلا به AD شناسایی کردند. با این وجود، در این بیماران، گرههای نوروفیبریلاری همزمان با A β و حاوی ژنهای خاص B. burgdorferi مانند OspA و فلاژلین بودند (Miklossy et al., 2004). در مطالعه دیگری (Fallon and Nields, 1994).دخالت B. burgdorferi در اختلالات تخریب عصبی از طریق ارتباط دمانس و میکروگلیوز با آتروفی قشر در بیماری لایم مشخص شد (Fallon and Nields, 1994). (Miklossy; 2015) 147 بیمار آلزایمر را برای جداسازی اسپیروکت ها بررسی کرد.گونه ها با کشت قشر مغز و خون خود بر روی محیط کشت اصلاح شده Noguchi و Barbor-Stoenner-Kelly II (BSK II). جدایه های باکتریایی با استفاده از میکروسکوپ الکترونی روبشی و میکروسکوپ نیروی اتمی برای وجود اندوفلاژلا، یعنی ویژگی منحصر به فرد اسپیروکت ها تجزیه و تحلیل شدند. این مطالعه اسپیروکتهایی را در خون، قشر مغز و CSF 14 بیمار مبتلا به AD شناسایی کرد که در موارد شاهد وجود نداشتند. علاوه بر این، پپتیدوگلیکان باکتریایی (PGN) با استفاده از آنتی بادی های خاص از طریق روش های ایمونوهیستوشیمی شناسایی شد. هیبریداسیون درجا نیز برای شناسایی DNA گونه خاص اعمال شد. PGN اختصاصی برای اسپیروکت ها در مغز ۳۲ بیمار AD و ۱۲ مورد با تغییرات خفیف قشر مغز شناسایی شد (McCoy et al., 2006). مطالعات هیستوپاتولوژی نشان داد که اسپیروکت ها در پیچ و تاب های عصبی فیبریلاری، پلاک های پیری و دیواره ناحیه قشری بیماران AD وجود دارند. شایان ذکر است که اسپیروکتها و آنتیژنهای اختصاصی آنها نیز با A β در مغز مرتبط بودند که نشاندهنده دخالت اسپیروکتها در زوال عقل و پاتوژنز AD است (Miklossy et al., ۲۰۰۴).
دادههای زیادی وجود دارد که نشان میدهد اسپیروکتها نوروتروپیسم قوی دارند و میتوانند سلولهای مغز را تحت تأثیر قرار دهند و باعث عفونتهای نهفته شوند. آنها همچنین می توانند از طریق لنفاوی، مجاری رشته های عصبی و عقده های سه قلو منتشر شوند (Riviere et al., 2002). این میکروارگانیسم ها می توانند از طریق tractus olfactorius نیز منتقل شوند. به طور جالبی نشان داده شده است که دستگاه بویایی در بیماران AD تحت تأثیر اسپیروکت ها قرار می گیرد. اسپیروکت ها دارای اجزای سطحی مختلفی مانند آمیلوئیدهای باکتریایی، پروتئین های اتصال به کلاژن و پروتئین های تشکیل دهنده منافذ هستند که به آنها در اتصال آنها به سطح سلول های میزبان کمک می کند (Brissette et al., 2010). مکانیسم تخریب عصبی آنها احتمالاً به دلیل آمیلوئیدهای باکتریایی است که باعث التهاب و اصلاح انعقاد خون از طریق فعال شدن پلاسمینوژن و فاکتور XII می شود. سلولهای میزبان، بهویژه، میکروگلیا و فاگوسیتها، اسپیروکتها را از طریق گیرندههایی که روی سطوح آنها قرار دارند، تشخیص میدهند. مهمترین گیرندههای تشخیص، گیرندههای Toll مانند (TLRs) هستند که در مغز نیز یافت میشوند (Crack and Bray, 2007). ماکروفاژها و میکروگلیاهای فعال شده در پاسخ به عفونتهای اسپیروکتالی که باعث التهاب میشوند، سیتوکینها و کموکاینها را تولید میکنند. به دنبال عفونت های اسپیروکتال، Aβ نیز در مغز انباشته می شود (شکل ۲B). علاوه بر این، سایر اجزای باکتریایی خاص مانند اسیدهای آمینه D و PGN نیز در مغز بیماران AD شناسایی شده است. قابل توجه است که گیرنده های تشخیص در سطح سلول های میزبان در مغزهای AD تنظیم مثبت فرض شده اند. در میان آنها، فعال شدن TLR2، TLR4 و TLR9 با تأثیر قابل توجهی بر مصرف در شرایط آزمایشگاهی Aβ گزارش شده است (Minoretti et al., 2006; Tahara et al., 2006). اسپیروکت ها می توانند سیستم ایمنی کلاسیک و جایگزین را با فعال کردن پاسخ های التهابی و آبشار لخته شدن منجر به نفوذپذیری عروقی کنند. پاسخ های التهابی ممکن است از طریق لیپوپروتئین های اسپیروکت (یعنی التهاب سیستماتیک) یا بیان بیش از حد فاکتور نکروز تومور ماکروفاژ (TNF) باشد. علاوه بر این، افزایش سطح سرمی آمیلوئید A (SAA) و پروتئین واکنشی C (CRP) در عفونتهای اسپیروکتال نشان داده شده است. اسپیروکت ها توانایی القای التهاب سیستمیک را دارند. جالب توجه است که میکروگلیاهای فعال در اطراف پلاک های پیری در مغز بیماران مبتلا به AD مشاهده شده است (McGeer and McGeer, 2002).
به طور کلی، اسپیروکت ها می توانند علائم بیولوژیکی، بالینی و پاتولوژیک AD مانند تجمع Aβ را تولید کنند. به طور خاص، ضایعات در سلولهای عصبی اولیه و گلیال و همچنین تودههای سلولهای مغزی پس از قرار گرفتن در معرض اسپیروکتها مشابه ضایعاتی هستند که در AD رخ میدهند (یعنی ضایعات پلاک مانند، درهمتنهمانند، ضایعات دژنراسیون گرانولوواکوئولار). بسیاری از مطالعات و بررسیها به ارتباط قوی بین AD و عفونت اسپیروکتال نتیجهگیری کردهاند که ۹ معیار هیل را در وجود یک رابطه علی برآورده میکند (Miklossy، ۲۰۱۱ ، ۲۰۱۵).
لیستریا مونوسیتوژنز
لیستریا مونوسیتوژنز یک باکتری گرم مثبت درون سلولی است که معمولاً به عنوان یک پاتوژن متولد شده از غذا شناخته می شود. L. monocytogenes یک عامل حدت به نام لیستریولیزین O (LLO) را مخفی می کند که در اختلالات تخریب عصبی نقش دارد. LLO یک سیتولیزین منفذ ساز است که فاگوزوم را پس از درونی شدن پاتوژن در سلول میزبان تجزیه می کند. این سم عضوی از خانواده سیتولیزین های وابسته به کلسترول (CDC) است که عمدتاً به صورت مونومرهای محلول ترشح می شود و الیگومریزه می شود تا یک پیش منافذ در سطح غشاء با مقدار کلسترول بالا ایجاد کند. تشکیل منافذ نتیجه تبدیل دو مارپیچ α به گیره های موی β است که غشاء را بیشتر تا ساختار دولایه گسترش می دهد تا یک بشکه β تولید کند.با کانال ۲۵ نانومتری به خوبی شناخته شده است که تجمع پروتئین یک نشانگر مهم در چندین اختلال تخریب عصبی مانند AD، PD و HD است که می تواند توسط لیستریا مونوسیتوژنز القا شود (شکل ۳). روشهای بیوشیمیایی و مولکولی وجود LLO را در سلولهای آلوده نشان دادهاند. اخیراً، ارتباط LLO ترشح شده به سلول ها با توده های بزرگ با استفاده از روش ایمونوفلورسانس نشان داده شده است (Viala et al., 2008). این سنگدانه ها سرشار از sequestosome1 یا p62 هستند و حاوی پروتئین های پلی یوبی کوئیتینه هستند. پروتئین p62 یک پروتئین آداپتور با دامنه PB1 در انتهای N همراه با یک دامنه یوبیکوئیتین است. پایانه N مسئول برهمکنش با زیر واحد S5a/Rpn10 پروتئازوم است در حالی که دامنه دیگر توانایی اتصال زنجیره های پلی یوبیکوئیتین را دارد. با توجه به این توانایی، به نظر می رسد که p62 به عنوان یک پروتئین آداپتور برای اتصال، ذخیره و ارائه پروتئین های ubiquitinated به پروتئازوم ها عمل می کند. (Viala et al., 2008) تجمعات LLO را در سیتوزول سلولهای آلوده با L. monocytogenes مطالعه کرده اند. آنها آنتیبادیهای مونوکلونال اختصاصی را برای تشخیص LLO در ماکروفاژهای مشتق از مغز، که با L. monocytogenes نوع وحشی آلوده بودند، اعمال کردند .. رویکرد ایمونوفلورسانس وجود LLO را در سلولها بهعنوان شکلهای دایرهای تولیدکننده سیگنال نقطهگذاری و گروههای بزرگ متراکم نشان داده است. سنگدانه های LLO حاوی پروتئین های پلی یوبی کوئیتینه با پروتئین میزبان p62 و سم LLO بودند. از آنجایی که تجمع پروتئینها با بیماریهای دژنراتیو مرتبط است، تجمع LLO در سلولها میتواند شبیه به تجمع پروتئینهایی باشد که با بیماریهای تخریب عصبی مرتبط هستند. علاوه بر این، فاگوسیت های تک هسته ای آلوده، TNFα را آزاد می کنند که باعث التهاب می شود. این باکتری حتی میتواند سلولهای اندوتلیال را مستقیماً آلوده کند یا از لکوسیتهای خون محیطی آلوده به سلولهای اندوتلیال سرایت کند و باعث تهاجم عصبی شود.
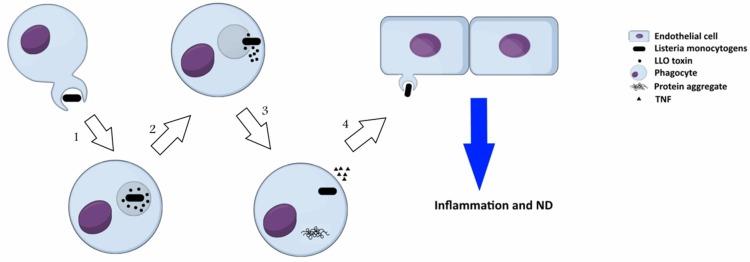 شکل ۳
شکل ۳ویروسها
رتروویروس ها گروهی از ویروس های حاوی RNA هستند که باعث ایجاد طیف وسیعی از بیماری های تخریب عصبی می شوند. بیماریهای نوروویروسی بهعنوان نوروتروپیسم، عفونت انتخابی نورونها توسط ویروسها و نورویرولانس یا بیماریهای عصبی ناشی از ویروس طبقهبندی میشوند. در اینجا، ما ویروس هایی را که در عصب کشی نقش دارند با تمرکز بر ویروس Epstein Bar (EBV)، ویروس هرپس سیمپلکس نوع ۱ (HSV-1)، ویروس نقص ایمنی انسانی نوع ۱ (HIV-1)، ویروس JC (JCV)، ویروس سرخک مورد بحث قرار می دهیم. (MV) و نوع C یا ویروس های انکوژنیک. مروری سریع بر نوروویرولانس این ویروس ها در جدول ۲ ارائه شده است.
جدول ۲
نورویرولانس برخی از ویروس ها در انسان
| گروه / جنس | خانواده | نورویرولانس |
|---|---|---|
| هرپس ویروس | ویروس اپشتین بار | ایجاد عفونت های نهفته و پایدار در سلول های B حافظه برای ذخیره ژنوم ویروسی. ترویج مونونوکلئوز عفونی و تحریک پاسخ خود ایمنی. افزایش خطر ابتلا به ام اس |
| ویروس سیمپلکس | HSV-1 | ایجاد آنسفالیت هرپس سیمپلکس در همان نقاط مغز که در بیماران AD ایجاد می شود. رمزگذاری گلیکوپروتئین B با تشابه توالی بالا به Aβ. کاهش تولید پردازش پروتئین پیش ساز آمیلوئید و القای تولید β – و γ – ترشحات برای تجمع Aβ. |
| لنتی ویروس ها | HIV-1 | درگیر کردن سیستم ایمنی از طریق عفونت سلول های CD4 + T و ماکروفاژها عبور از سد خونی مغزی با ماکروفاژها. توانایی تکثیر در سلول های غیرقابل تقسیم مانند ماکروفاژها یا سلول های تقسیم شونده مانند میکروگلیا. تحریک التهاب از طریق القای آزادسازی کموکاین ها، سیتوکین ها و اسیدهای آمینه تحریکی پروستاگلاندین ها از سلول های آلوده. ایجاد اختلالات عصبی شناختی مرتبط با HIV در قسمت های مختلف مغز. کاهش حجم ساختارهای مغز و ایجاد ناهنجاری در ماده سفید مغز. افزایش سطح نوروفیلامنت ها و افزایش انتشار پلاک های Aβ و تجمع بعدی آن در هیپوکامپ و لوب فرونتال. |
| پلیوماویریده | ویروس JC | ایجاد عفونت نهفته در قسمت های مختلف بدن مانند سیستم عصبی مرکزی، روده و کلیه. بروز مجدد عفونت در افراد دارای نقص ایمنی رندر آنسفالوپاتی JCV، نورونوپاتی سلول گرانول JCV، مننژیت JCV و لوکوآنسفالوپاتی چند کانونی پیشرونده. |
| موربیلی ویروس | ویروس سرخک | سلول های لنفوسیتی را آلوده می کند و سپس در غدد لنفاوی تکثیر می کند. تحریک یک لکوپنی قوی عبور از سد خونی مغزی با لکوسیت ها یا آلوده کردن سلول های اندوتلیال ریز عروقی برای حمله به مغز از طریق مهاجرت از شبکه مشیمیه یا پیاز بویایی. القای آنسفالیت حاد پس از عفونی سرخک یا پانانسفالیت اسکلروزان تحت حاد پس از دوره سرخک. |
| ویروس های انکوژنیک / نوع C | HTLV-1 | بدخیمی یا سندرم عصبی در انسان. |
| HTLV-2 | HTVL-1 باعث لوسمی/لنفوم سلول T بزرگسالان می شود. اختلالات التهابی مانند میلوپاتی پیشرونده یا میلوپاتی مرتبط با HTLV-1. | |
| MuLV | آستروسیت ها، سلول های اندوتلیال مغز، لکوسیت ها، میکروگلیا و الیگودندروسیت ها را آلوده می کند. ایجاد تغییرات اسفنجی شکل در نخاع و ساقه مغز و همچنین نقص های رفتاری مانند فلج اندام عقبی و لرزش. فعال کردن سلول های گلیال و افزایش ترشح مواد پیش التهابی. ترویج مرگ سلول های عصبی |
ویروس اپشتین بار
عفونت EBV عمدتاً در دوران کودکی و بدون علائم ظاهری رخ می دهد. با این حال، می تواند با علائم بالینی مانند مونونوکلئوز عفونی، به ویژه در بزرگسالان ظاهر شود. تخمین زده شده است که تقریباً ۹۰ درصد افراد در سراسر جهان به EBV آلوده هستند. به طور کلی پذیرفته شده است که EBV با برخی از بیماری های خودایمنی مانند تومورهای انسانی و MS (تا ۹۹٪) مرتبط است. این سازگاری را میتوان با توانایی این ویروس در ایجاد عفونتهای نهفته و پایدار در سلولهای B حافظه برای ذخیره ژنوم ویروس توضیح داد. جالب توجه است که مونونوکلئوز عفونی ناشی از EBV می تواند به طور قابل ملاحظه ای خطر ابتلا به ام اس را افزایش دهد زیرا افراد دارای آنتی ژن هسته ای ضد EBV (EBNA-1) IgG و آنتی بادی های ضد VCA IgG دارای خطر افزایش یافته MS هستند (Santiago et al., 2010) افزایش آنتی بادی های ضد EBNA-1 منجر به واکنش متقابل با آنتی بادی های خود در بیماران ام اس می شود. نشان داده شده است که EBNA-1 دارای واکنش متقاطع با ریبونوکلئوپروتئین هسته ای ناهمگن L (HNRNPL) است که HNRNPL را به عنوان یک آنتی ژن معرفی می کند (Lindsey et al., 2016). واکنش متقاطع آنتی بادی های اختصاصی پروتئین پایه میلین مونوکلونال (MBP) به دست آمده از بیماران ام اس با پروتئین غشای نهفته ۱ (LMP1) EBV نمونه دیگری از پاسخ خود ایمنی در MS است. به طور خاص، تزریق LMP1 به موش باعث القای اتوآنتی بادیهای واکنشدهنده میلین میشود (Lomakin et al., 2017). همانطور که قبلا ذکر شد، اکثر افراد بالغ آلوده به EBV علائم ام اس را نشان نمی دهند، که نشان می دهد EBV به تنهایی برای ایجاد ام اس کافی نیست. با این حال، ترکیب آن با سایر عوامل از جمله نوع EBV، کمبود ویتامین D و حساسیت ژنتیکی سیستم ایمنی مانند ژن MHC کلاس II میتواند باعث ایجاد ام اس شود (Najafipoor et al., 2015).
در مورد وجود EBV در مغز بیماران ام اس و ارتباط آن با پاتوژنز ام اس اختلاف نظر وجود دارد. برخی از مطالعات اشاره کرده اند که این ویروس در سلول های B مغز و در ساختارهای فولیکول مانند مننژ وجود دارد، در حالی که گزارش های دیگر بیان کرده اند که هیچ نشانه ای از EBV وجود ندارد یا تنها جمعیت کمی از سلول های B آلوده به ویروس در مغز وجود دارد. بیماران ام اس تورکیلدسن و همکاران (Torkildsen et al., 2010) ضایعات ماده خاکستری قشر بیماران ام اس را بررسی کرد و به جای تجمع سلول های B، تنظیم مثبت ژن های مرتبط با ایمونوگلوبولین را در این بخش ها توسط سلول های پلاسما مشاهده کرد. علاوه بر این، آنها نتوانستند شواهدی از عفونت فعال یا نهفته EBV را شناسایی کنند. این یافته ممکن است به این دلیل باشد که ضایعات ماده خاکستری از نظر پاتولوژیک با ضایعات ماده سفید متفاوت است که نشان دهنده فقدان یا کمبود سلول های ایمنی نفوذی از جمله لنفوسیت ها است. در مطالعه دیگری، (Willis et al., 2009) از PCR بلادرنگ برای تشخیص ژنوم ویروسی و تقویت RNA کدگذاری شده با EBV از ضایعات ماده سفید ۱۷ بیمار MS استفاده کرد. هیچ نتیجه مثبتی مشاهده نشد به جز سیگنالی که بیانگر بیان RNA پیام رسان CD20 و حضور +CD20 است. سلول های B. علاوه بر این، مجموعه دیگری از نمونهها شامل تودههای سلول B در پارانشیم و نفوذ سلولهای B آزاد در مننژها مورد بررسی قرار گرفت و مقادیر بسیار کمی از EBV RNA را تنها در دو مورد از ۱۲ نمونه بیماران MS نشان داد. در مقابل، تشخیص RNA کوچک کدگذاری شده با EBV (EBER) با استفاده از روش هیبریداسیون درجا، وجود EBV را در مغز بیماران ام اس تایید کرد (Serafini et al., 2007). EBER1 و EBER2 دو رونوشت ترجمه نشده از EBV هستند که عمدتاً در هسته سلول قرار دارند و در عفونت پنهان ویروس بیان می شوند. با استفاده از این روش، تعداد زیادی سلول EBER مثبت از ماده سفید و مننژ افراد مبتلا به MS یافت شده است که بیشتر در فولیکول های سلول B بودند (Serafini et al., 2007).
به طور کلی، مشارکت مستقیم عفونت EBV در ایمونوپاتولوژی ام اس بعید است. با این حال، ممکن است از طریق تأثیر غیرمستقیم بر عملکرد ایمنی یا از طریق تقلید مولکولی بین آنتیژنهای CNS و EBV به MS کمک کند (Willis et al., 2009).
ویروس هرپس سیمپلکس نوع ۱
عفونت HSV-1 معمولاً یک عفونت پوستی به نام زخم سرد ایجاد می کند که در سنین اولیه افراد رخ می دهد و در محل آلوده اولیه (بیشتر در لب) شروع به تکثیر می کند. پس از آن، ویروس به سلول های عصبی حرکت می کند و وارد فاز نهفتگی در سیستم عصبی محیطی می شود. در این مرحله، ژنوم ویروس را می توان یافت اما پروتئین های ویروسی غیرقابل شناسایی هستند. هنگامی که ویروس از مرحله نهفتگی خارج می شود، شروع به تکثیر و تولید پروتئین های ویروسی و ویریون های کامل می کند که منجر به عفونت حاد می شود. HSV-1 باعث ایجاد انسفالیت هرپس سیمپلکس (HSE) در همان نقاط مغز می شود که در بیماران AD. علاوه بر این، HSV-1 ممکن است به دلیل توانایی ویروس برای پنهان ماندن طولانی مدت در سلول های عصبی و همچنین وجود شکل فعال ویروس در تعداد زیادی از افراد مسن، در AD دخیل باشد. مشابه آلزایمر،APOE-ɛ۴ (Wozniak et al., 2009).
HSV-1 می تواند گلیکوپروتئین B را با شباهت و ویژگی های توالی بالا به Aβ رمزگذاری کند و پردازش پروتئین پیش ساز آمیلوئید (APP) را کاهش می دهد. به طور خاص، سلولهای عصبی آلوده به HSV-1 سطح پایینتری از APP، افزایش زیادی در قطعه kDa 55 APP و سطوح بالاتر Aβ درون سلولی را نشان دادند. علاوه بر این، HSV-1 باعث تولید آنزیمهایی شد که مسئول تشکیل Aβ ، β- و -سکرتازها هستند، که Aβ را در سلولهای آلوده به HSV-1 و مغز موشها جمعآوری میکنند که منجر به پلاکهای آمیلوئید میشود (Wozniak et al., 2009). بر این اساس، این پاتوژنز HSV-1 این ویروس را به عنوان یک عامل بالقوه اتیولوژیک مهم در AD معرفی می کند.
ویروس نقص ایمنی انسانی نوع ۱
لنتی ویروس ها از اعضای مهمی شامل ویروس های نقص ایمنی مختلف گاو (BIV)، گربه (FIV)، انسان (HIV)، سیمین (SIV) و ویروس visna-maedi (VMV) تشکیل شده اند. اگرچه لنتی ویروس ها ژن های لنتی ویروسی متنوعی را در بر می گیرند، اما حاوی ویژگی های ژنتیکی و بیولوژیکی مشترکی مانند سازماندهی ژنوم رتروویروسی هستند که برای حدت آنها ضروری است. آنها توانایی تکثیر در سلول های غیرقابل تقسیم مانند ماکروفاژها را دارند. علاوه بر این، لنتی ویروس ها اغلب ترجیح می دهند ماکروفاژها و میکروگلیاها را به جای آستروسیت ها و سلول های اندوتلیال آلوده کنند. مکانیسم نوروپاتوژنیک در لنتی ویروس ها از طریق عفونت ماکروفاژهایی است که با عبور از سد خونی مغزی وارد CNS می شوند. گیرنده های سطح سلولی برای برخی از لنتی ویروس ها (مانند HIV و SIV) در داخل و خارج از سیستم عصبی مانند CD4 شناسایی شده اند، chemokine receptors and CC chemokine receptor 5.(CCR5; Power, 2001). آسیب شناسی عصبی لنتی ویروس ها با عوامل متعددی مانند سن میزبان و وضعیت سیستم ایمنی آن تعیین می شود. همچنین بستگی به سویه لنتی ویروس فردی دارد. بیماران آلوده به لنتی ویروس دچار زوال عقل یا انسفالوپاتی همراه با التهاب و آسیب سلول های عصبی می شوند. عقده های قاعده ای و قشر پیشانی به عنوان محل های اصلی نفوذ و عفونت ماکروفاژها شناخته شده اند و سلول های عصبی این قسمت ها را در برابر عفونت های لنتی ویروسی آسیب پذیر می کنند. عفونت های ناشی از لنتی ویروس به شدت سلول های عصبی را تغییر می دهند و باعث واکوئل شدن اندریتیک، آسیب آکسون، از دست دادن نورون همراه با تغییر در نفوذپذیری سد خونی مغز و عملکرد آن می شوند (Everall et al., 1999).
عفونت سلول های مغز با HIV-1 می تواند منجر به التهاب عصبی، اختلال عملکرد حرکتی و سندرم زوال عقل HIV شود. HIV-1 عمدتاً ماکروفاژها و میکروگلیاها را آلوده می کند که منجر به آزاد شدن کموکاین ها، سیتوکین ها و اسیدهای آمینه تحریکی پروستاگلاندین ها از سلول های آلوده می شود. این عوامل آزاد شده باعث التهاب و آسیب عصبی می شوند. بر اساس گرایش HIV به سلول های انسانی، HIV-1 به گونه های M- و T-tropic طبقه بندی می شود. ماکروفاژها توسط سویه های M-tropic HIV-1 از طریق گیرنده کموکاین CC 5 (CCR5) مورد هدف قرار می گیرند، در حالی که سویه های T-tropic از گیرنده CXC 4 همراه با پروتئین G (CXCR4) برای آلوده کردن ماکروفاژها استفاده می کنند. کموکاین SDF-1 یک مولکول مهم است که می تواند CXCR4 را متصل کند و این گیرنده را برای اتصال HIV-1 در دسترس نباشد.(Albright and González-Scarano, 2004).
ویروس این قابلیت را دارد که عمدتاً سیستم ایمنی را با عفونت سلول های CD4 + T و ماکروفاژها درگیر کند. قبل از یافتن یک درمان موثر، ۸ تا ۱۵ درصد از بیماران مبتلا به عفونت HIV از یک بیماری شناختی شدید به نام زوال عقل مرتبط با HIV (HAD) رنج میبردند. این تعداد پس از توسعه درمان ترکیبی ضد رتروویروسی به ۲ درصد کاهش یافته است (cART; Heaton et al., 2010). با این حال، استفاده از دوز cART ویروس را به طور کامل از سلول های مغز حذف نمی کند، بنابراین بیماران مبتلا به HIV ممکن است عفونت مزمن در CNS داشته باشند. قابل توجه است که اکثر داروهای ضد ویروسی نمی توانند به طور موثر به مغز نفوذ کنند. بنابراین، عفونت ویروسی همچنان در CNS پیشرفت می کند. اختلالات عصبی-شناختی مرتبط با HIV (HAND) به اختلالات عصبی شناختی از جمله کمپلکس زوال عقل ایدز، آنسفالیت HIV، انسفالوپاتی HIV و اختلال حرکتی شناختی جزئی اشاره دارد که ویژگی های بالینی مختلفی را نشان می دهد. پاتوفیزیولوژی دست شامل التهاب و اختلال عملکرد سلول های عصبی است. مطالعات تصویربرداری و بررسی CSF نشان داده است که التهاب در عفونت HIV در عرض ۱۸ روز پس از قرار گرفتن در معرض ویروس رخ می دهد (Valcour et al., 2012) از طریق فعال شدن ماکروفاژها منجر به اختلال CNS می شود. ماکروفاژهای آلوده به HIV میکروگلیا را فعال میکنند و منجر به سلولهای غولپیکر با چند هسته و انتشار آستروگلیوز میشوند. در نتیجه، این پاسخ در انسفالوپاتی HIV، از بین رفتن سلول های عصبی و تخریب عصبی در قسمت های مختلف مغز نقش دارد. طیفسنجی رزونانس مغناطیسی تغییراتی را در مقدار متابولیتهای مغز بیماران HIV شناسایی کرده است. این اثر با التهاب، گلیوز و از دست دادن سلول های عصبی همراه است و حتی در افراد تحت درمان با cART نیز مشاهده شده است (Harezlak et al., 2011). تصویربرداری رزونانس مغناطیسی نشان داد که حجم ساختارهای مغز در افراد مبتلا به HAD کاهش یافته است. علاوه بر این، تصویربرداری تانسور انتشار، یک فناوری مفید برای اندازهگیری انتشار مولکولهای آب، ناهنجاریهایی را در ماده سفید مغز HAD بیماران نشان داده است. درمان cART ممکن است ناهنجاری ها را بهبود بخشد اما وضعیت طبیعی را به طور کامل باز نمی گرداند. علاوه بر این، اندازهگیری فعالیت متابولیک در مغز با استفاده از فناوری توموگرافی انتشار پوزیترون، مناطقی را در مغز HAD با متابولیسم تغییر یافته نشان داده است که با وضعیت شناختی مرتبط است (von Giesen et al., 2000).
نوروفیلامنت ها (NFs)، به عنوان مثال، پروتئین های عصبی خاص با وزن مولکولی کم، متوسط و بالا ممکن است در جریان خون و CSF در نتیجه از دست دادن یا اختلال نورون ها یافت شوند. این به عنوان یک نشانگر زیستی مهم در اختلالات تخریب عصبی مانند AD، ASL، MS و دمانس عروقی زیر قشری در نظر گرفته می شود (Malmeström et al., 2003; Pasol et al., 2010). قابل توجه است که سطوح NFs در CSF بیماران HIV افزایش می یابد و در افراد HAD به طور قابل توجهی بالاتر است. نشان داده شده است که درمان cART می تواند سطوح NFs را کاهش دهد و به دنبال آن اختلالات شناختی را بهبود بخشد (Jessen Krut et al., 2014). علاوه بر این، آسیب عصبی با ماکروفاژها و فعال شدن مونوسیت ها در ارتباط است. و neopterin، نشانگر زیستی فعال شدن ماکروفاژها و مونوسیت ها، در CSF بیماران HAD شناسایی شده است. CD14 متصل به غشاء، یک گیرنده برای لیپوپلی ساکارید، نشانگر فعال شدن مونوسیت ها است که شکافته شده و به شکل محلول، یعنی sCD14 تبدیل می شود. اندازه گیری سطوح sCD14 پلاسما ارتباط بین این نشانگر زیستی و اختلال شناختی عصبی در بیماران HIV را نشان داد (Lyons et al., 2011). علاوه بر این، یکی دیگر از نشانگرهای فعال سازی ماکروفاژها/مونوسیت ها، CD163، در بیماران مبتلا به HIV با اختلالات شناختی افزایش می یابد (Burdo et al., 2013).
مشخصه AD، به عنوان مثال، تجمع β در بیماران HIV نیز رخ می دهد. در بیماران HIV پلاک های β منتشر شده و متعاقباً در هیپوکامپ و لوب فرونتال تجمع می یابند. مطالعات پاتولوژیک نشان داد که تجمع β در مغز بیماران مبتلا به AD معمولاً در نواحی نئوکورتیکال رخ می دهد.
اگرچه، بسیاری از داده ها همبستگی قوی بین تجمع Aβ در مغز و AD را برجسته می کنند، تنها تعدادی از مطالعات این ناهنجاری را به عنوان عامل اصلی در آسیب شناسی دست نشان می دهند (Cohen et al., 2015). HIV میتواند نفوذپذیری سد خونی مغزی را از طریق اختلال در یکپارچگی سلولهای اندوتلیال میکروواسکولار و اتصالات سلولی محکم ایجاد کند، که دسترسی ماکروفاژهای آلوده به مغز را تسهیل میکند. اختلال یکپارچگی سد خونی مغزی بر تجمع Aβ در مغز بیماران HIV به دلیل ناتوانی آن در فیلتر کردن پپتید Aβ تأثیر می گذارد که نشان دهنده ویژگی های عصبی آسیب شناختی مشابه AD و HIV است (Erickson and Banks, ۲۰۱۳).
ویروس JC
JCV یک ویروس DNA دو رشته ای دایره ای نوروتروپیک متعلق به Polyomavirideae است. این ویروس تا ۷۰ درصد شباهت ژنومی با ویروس BK و SV40 دارد. ژنوم آن از سه بخش اصلی شامل یک منطقه کدگذاری اولیه، یک منطقه کدگذاری دیررس و یک منطقه تنظیمی تشکیل شده است. این ویروس معمولا باعث عفونت نهفته در قسمت های مختلف بدن مانند CNS، روده و کلیه می شود. ظهور مجدد عفونت های JCV از جمله انسفالوپاتی JCV (JCVE)، نورونوپاتی سلولی گرانول JCV (GCN)، مننژیت JCV، و لوکوآنسفالوپاتی چند کانونی پیشرونده (PML) اغلب در افراد دچار نقص ایمنی با انواع مختلف بیماری ها رخ می دهد.
PML یک بیماری دمیلینه کننده خطرناک ناشی از ویروس JC در بیماران مبتلا به نقص ایمنی است. مشخصه بیماری عفونت الیگودندروسیت ها با وجود هسته های بزرگ شده و انکلوزیون های داخل سلولی است. سلولهای آلوده معمولاً در قسمتهای محیطی ضایعات دمیلینهکننده به خوبی مشخص شده یا مناطق مختل در موارد رنگ پریدگی میلین مشاهده میشوند. آسیب شناسی عصبی PML با علائم بالینی متنوعی مانند ضایعات دمیلینه کننده در ماده سفید زیر قشر مغز و ناحیه قشر مغز همراه است (Moll et al., 2008). احتمالاً بیشترین ناحیه آسیب دیده لوب های فرونتال و قسمت های پاریتواکسیپیتال و به میزان کمتری در مخچه، ساقه مغز و نخاع بیماران PML است (Bernal-Cano et al., 2007). شدت دمیلینه شدن سلول ها از رنگ پریدگی میلین تا دمیلینه شدن همراه با آسیب آکسون و ماکروفاژهای حامل بقایای میلین و ایجاد ضایعات سوخته که اغلب در بیماران مبتلا به ایدز دیده می شود متفاوت است (Gray et al., 2003).
GCN یکی دیگر از بیماری های مرتبط با JCV است که با عفونت لیتیک در حال ظهور نورون های سلولی گرانول که منجر به از دست دادن نورون ها و آتروفی مخچه می شود مشخص می شود. بیشتر موارد با GCN در بیماران HIV گزارش شده است (Agnihotri et al., 2014). تخریب عصبی در برخی از بیماران مبتلا به GCN با یافته های تصویربرداری مشاهده شده است. تغییرات ماده سفید به خصوص در دمگل های مخچه میانی و پونز در ارتباط با ماده خاکستری در این بیماران مشاهده شده است. علاوه بر این، مستند شده است که GCN می تواند در آستروسیت ها و الیگودندروسیت ها رخ دهد (Wijburg et al., 2015).
JCVE به عنوان یک سندرم نورودژنراتیو جدید ناشی از JCV در سال ۲۰۰۹ معرفی شد. این بیماری با شناسایی ضایعات محدود به ماده خاکستری و وجود ویروس در ماده خاکستری قشر مغز و همچنین CSF بیماران مشخص می شود. JCVE عمدتا بر نورون های هرمی مغز و آستروسیت ها در ماده خاکستری نیمکره تأثیر می گذارد. وجود ماکرومولکول های ویروسی مانند پروتئین های ویروسی در نورون های آکسون گزارش شده است که نشان دهنده مهاجرت JCV به مغز از طریق عفونت آکسون های نورون ها است (Miskin and Koralnik، ۲۰۱۵).
ویروس سرخک
MV، عضوی از جنس Morbillivirus، به عنوان یک ویروس بسیار مرتبط با سلول شناخته می شود. ویروس از طریق دستگاه تنفسی وارد بدن میزبان می شود و به سلول های لنفوسیتی حرکت می کند، سپس در غدد لنفاوی تکثیر می شود. در مراحل اولیه عفونت، به راحتی می توان آن را از لکوسیت های خون جدا کرد. یک لکوپنی قوی در بیماران سرخک به دلیل افزایش اتصال لنفوسیت ها به اندام های لنفاوی رخ می دهد. پس از تکثیر ویروس در اندام های لنفاوی، می توان آن را در بافت ها و سلول های دیگر مانند پوست، سلول های اندوتلیال و مغز یافت. دسترسی به سلولهای مغز از راههای مختلفی از جمله انتقال لکوسیتهای آلوده از طریق سد خونی مغزی، عفونت سلولهای اندوتلیال میکروواسکولار و مهاجرت از طریق شبکه مشیمیه یا حباب بویایی صورت میگیرد (McQuaid and Cosby, 2002). افزایش چسبندگی لکوسیت ها در طول عفونت MV ظرفیت مهاجرت سلول ها را از طریق موانع اندوتلیال کاهش داد و عفونت سلول های اندوتلیال را افزایش داد (Dittmar et al., 2008).
پس از دوره سرخک، MV می تواند دو شکل مختلف عفونت را در CNS ایجاد کند: آنسفالیت حاد سرخک پس از عفونی (APME) یا پانانسفالیت اسکلروزان تحت حاد (SSPE). تخمین زده شده است که AMPE، شایع ترین عارضه CNS، در ۰.۱٪ از بیماران مبتلا به MV با نرخ مرگ و میر ۲۰٪ مشاهده می شود. بر اساس داده های الکتروانسفالوگرام، ۵۰ درصد بیماران از اختلال عملکرد مغزی رنج می برند. وجود اسید نوکلئیک MV تنها در CNS بیماران APME شناسایی شده است. بنابراین، علائم بالینی ممکن است مربوط به پاسخ خود ایمنی میزبان باشد. پاسخ التهابی ممکن است توسط سلولهای اندوتلیال آلوده مغز یا لکوسیتهای متصل به سلولهای اندوتلیال میکروواسکولار مغز آغاز شود (Reuter and Schneider-Schaulies, 2010). در مقابل، SSPE یک بیماری نادر و پیشرونده تخریب عصبی است که پس از سال ها تداوم ویروسی رخ می دهد. نوع MV جدا شده از CNS این بیماران با ویروس نوع وحشی متفاوت است. اولی کمبود است و ساختار ژنوم آن دارای جهش های مختلفی است که عمدتاً از طریق جایگزینی یوریدین توسط سیتیدین در ژن ماتریکس (ژن M) ایجاد می شود. مطالعات in vitro و in vivo نشان داده اند که علیرغم جهش های بیش از حد در ژن M، ویروس جهش یافته توانایی آلوده کردن مغز موش های بالغ و کشت سلول های عصبی اولیه را دارد (Patterson et al., 2001). به طور کلی، دلیل تداوم MV در CNS نامشخص است و کمبود بیماران SSPE این خط بررسی را پیچیده تر کرده است. از سوی دیگر، در نورون های انسانی، گیرنده های شناخته شده برای MV (یعنی نکتین ۴ و CD 150) بیان نمی شوند و مکانیسم انتشار MV در این سلول ها هنوز نامشخص است. گسترش واریانت MV در نورونها اساساً نیازمند تغییرات هیپرفیوزوژنیک بیثباتکننده در اکتودومین پروتئین F است که به ویروس اجازه میدهد جهت نورونها تروپیسم را نشان دهد (Watanabe et al., 2018).
ویروسهای انکوژنیک یا نوع C
ویروسهای انکوژنیک گروهی از رتروویروسها هستند که باعث بیماریهای عصبی در انسان و برخی حیوانات مانند پرندگان، گربهها و جوندگان میشوند. ژنوم این ویروس ها شامل سه ژن ساختاری اصلی شامل دو قسمت LRT و یک ژن کمکی است. ویروسهای لوسمی پرندگان (ALV)، ویروسهای لنفوتروپیک سلول T انسانی نوع ۱ و ۲ (HTLV-1 و HTLV-2) و ویروسهای لوسمی موشی (MuLV) گروههای اصلی رتروویروسهای انکوژنیک نوروتروپیک هستند.
HTLV-1 و HTLV-2
HTLV ها ویروس های پایداری هستند که می توانند به طور موثر لنفوسیت های T را تبدیل کرده و آنها را جاودانه کنند. در مقایسه با HTLV-1، هر دو سلول CD4+ T و CD8+ T حساسیت مشابهی نسبت به HTLV-2 دارند. با این حال، ویروس دوم بسیار کمتر شایع و بیماری زا است. به طور خاص، HTLV-1 ترجیحاً تروپیسم تغییر شکل را برای سلول های CD4+ T نشان می دهد، در حالی که HTLV-2 بار پروویروسی بالاتری را در سلول های CD8+ T نشان می دهد. بدخیمی یا سندرم عصبی (میزان بروز <1%) نمونه ای از اختلالات عصبی ناشی از ویروس HTLV در انسان است. HTLV-1 و HTLV-2 باعث ایجاد میلوپاتی پیشرونده می شوند که با التهاب در قسمت قفسه سینه نخاع تعریف می شود (Power, ۲۰۰۱). تخمین زده شده است که HTLV ها حدود ۲۰ میلیون نفر را در سراسر جهان مبتلا می کنند. افراد آلوده ممکن است بدون علامت یا عصبی باشند. هر دوی آنها به طور بالقوه می توانند ویروس را منتقل کنند.
HTLV-1 باعث یک بدخیمی لنفوپرولیفراتیو می شود که به آن لوسمی/لنفوم سلول T بالغ می گویند. علاوه بر این، ویروس می تواند باعث ایجاد اختلالات التهابی مانند میلوپاتی مرتبط با HTLV-1 (HAM/TSP) در کودکان و افراد زن شود. یافته های اولیه در مورد آسیب شناسی HAM/TSP اثر لنفوسیت های T سیتوتوکسیک CD4+ HTLV-1 Tax را نشان داد. آسیب پارانشیمی موضعی با آزادسازی سیتوکین ها و لنفوکین ها در طول فرآیند تبدیل سلول T امکان پذیر است. پاسخ خودایمنی میزبان یکی دیگر از پارامترهای مهم است که در طول پاتوژنز HTLV-1 در بیماران HAM/TSP ایجاد می شود. این بیماران آنتی بادی های IgG تولید می کنند که هم با HNRNP-A1 و هم با یک اپی توپ غالب ایمنی در Tax واکنش متقابل دارند (Levin et al., 2002).
اولین گزارش عفونت ناشی از HTLV-2 مربوط به بیماران مبتلا به هر دو عفونت HTLV-1 و HTLV-2 و میلوپاتی بود. مشخص شده است که برخی از بیماران با علائم HAM/TSP با عفونت منفرد HTLV-2 وجود دارند که نشان دهنده نقش مستقیم HTLV-2 در اختلال HAM/TSP است.
ویروسهای لوکمی موشی
Murine Leukaemia Viruses
MuLVها به عنوان بزرگترین گروه از رتروویروس های نوروتروپیک شناخته می شوند که باعث ایجاد برخی نقص های رفتاری مانند فلج اندام عقبی و لرزش می شوند. این نوع از رتروویروس ها اغلب آستروسیت ها، میکروگلیاها و الیگودندروسیت ها را آلوده می کنند. با این حال، برخی از گونههای MuLV وجود دارند که سلولهای عصبی را آلوده میکنند. سویههای MuLVs از گیرندههای سطح سلولی مختلفی برای اتصال سلولها مانند mCAT-1 استفاده میکنند، اگرچه هیچ مدرکی دال بر نقش این گیرنده در سیستم عصبی وجود ندارد. عفونت لکوسیت ها و سلول های اندوتلیال مغز توسط MuLVs محتمل ترین راه برای دسترسی به مغز است. اگرچه نوع آسیب شناسی عصبی ناشی از این ویروس ها به فشار بستگی دارد، تغییرات اسفنجی شکل در نخاع و ساقه مغز به عنوان شایع ترین علل آسیب شناسی شناخته شده است.ژن های gag یا env، با پاسخ میزبان. دو حوزه در ژن env Fr-98 MuLV نورویرولانس را در مدل موش کنترل میکنند و باعث رفتار غیرطبیعی و مرگ میشوند (Poulsen et al., 1998). اگرچه شواهد کمی برای پاسخ های میزبان بیماری زا در عفونت MuLV وجود دارد، فعال شدن سلول های گلیال و افزایش تولید پیش التهابی به عنوان مسیرهای اصلی نوروویولانس در نظر گرفته می شود. علاوه بر این، عفونت های MuLV باعث افزایش غلظت گلوتامات در محیط خارج سلولی از طریق تغییر در نفوذپذیری سد خونی مغزی می شود که منجر به مرگ سلول های عصبی می شود (Sei et al., 1998).
قارچها و مخمرها
ردپای قارچ در بیماران AD
اخیراً نقش قارچ ها در بیماری های تخریب عصبی توسط تعدادی از مطالعات مورد توجه قرار گرفته است. بر این اساس، پروتئین های قارچی، DNA و پلی ساکاریدها در خون و CSF بیماران AD یافت شده است. علاوه بر این، سلولهای قارچی مستقیماً در بخشهای مختلف CNS در افراد مبتلا به AD از جمله قشر فرونتال، نیمکره مخچه، قشر آنتورینال/هیپوکامپ و شبکه کوروئید با استفاده از ایمونوهیستوشیمی و میکروسکوپ کانفوکال شناسایی شدهاند. علاوه بر این، DNA قارچی، پروتئین و پلی ساکارید در سرم خون بیماران AD مشاهده شد. گونههای قارچی موجود در نمونههای CNS بیماران مبتلا به AD متعلق به گونههای Cladosporium ، Malassezia، Neosartorya، Phoma، Saccharomyces، Sclerotinia و Candida بودند (Pisa et al., 2015). علاوه بر این، آلونسو و همکاران. (Alonso et al., 2014) DNA و پروتئین های قارچی را در CSF با استفاده از روش PCR و اسلات بلات شناسایی کردند.
عفونتهای قارچی در بیماران ام اس
تصور میشود که خودایمنی در اماس توسط پروتئینها یا آنتیژنهای عجیب و غریب ایجاد میشود که مولکولهای خود را تقلید میکنند که منجر به واکنش علیه میلین و به دنبال آن تخریب سلولهای گلیال و عصبی میشود. وجود ماکرومولکول های قارچی مانند پروتئین ها و DNA در CSF بیماران ام اس توسط Pisa و همکارانش مورد مطالعه قرار گرفته است. (Pisa et al., 2015). آنها نشان داده اند که پروتئین های قارچی را می توان در CSF چندین بیمار ام اس تشخیص داد در حالی که DNA قارچی در برخی از نمونه ها تکثیر شده است. نتایج PCR وجود گونه های مختلف قارچی را در نمونه های CSF بیماران ام اس نشان داده است.
شواهدی وجود دارد که برخی از عفونت های قارچی را با ام اس مرتبط می کند. جنس کاندیدا به عنوان یک مخمر بیماری زا برای انسان به خوبی شناخته شده است که باعث ایجاد ضایعات دمیلینه در CNS بیماران آلوده به گونه های کاندیدا می شود (Lipton et al., 1984). از آنجایی که گونههای کاندیدا بهعنوان ارگانیسمهای مشترک در چندین قسمت از بدن انسان مانند دستگاه گوارش، حفره دهان و پوست یافت میشوند، آنتیبادیهایی علیه این گونهها در افراد عادی یافت میشود. با این حال، رویکرد ایمونوفلورسانس غیرمستقیم، تیتر آنتیبادیهای قابل توجهی را علیه گونههای کاندیدا در بیماران اماس نشان داده است (Benito-León et al., 2010). این فرضیه وجود دارد که قارچ ها و مخمرها از نظر بالینی بیماری زا مانند گونه هایی ازآسپرژیلوس و کاندیدا توانایی پوشاندن خود را از سیستم ایمنی با استفاده از پوشش مانوپروتئین خود دارند که به آنها کمک می کند به بافت های غیر عصبی بچسبند و سموم خود (مایکوتوکسین ها) را به جریان خون آزاد کنند (Purzycki and Shain, 2010). مایکوتوکسینها از مغز سد خونی عبور میکنند و بر آستروسیتها و الیگودندروسیتها تأثیر میگذارند که نقش مهم آنها حفظ یکپارچگی سد خونی-مغزی و حمایت غذایی از میلین است. در نتیجه، این سموم میلین را مستعد تخریب توسط عوامل مختلف میکنند که مشخصه اصلی ام اس است (Purzycki and Shain, 2010). فوزاریومگونه ها نوعی مایکوتوکسین به نام فومونیزین B تولید می کنند که در بیوسنتز اسفنگولیپیدها دخالت می کند. از بین رفتن اسفنگولیپیدها در ماده سفید یک علامت مشخصه بیماران ام اس است (Stockmann-Juvala and Savolainen, 2008). علاوه بر این، فومونیزین B اثر سمی بر آستروسیت های اولیه و میکروگلیای موش دارد (Stockmann-Juvala and Savolainen، ۲۰۰۸). پنیترم A توسط Penicillium crustosum تولید می شود که می تواند در اختلالات عصبی مختلف مانند آتاکسی، تورم میتوکندری، نیستاگموس و فلج کاذب در مدل های حیوانی دخیل باشد (Cavanagh et al., 1998).
گلیوتوکسین از گونههای آسپرژیلوس و کاندیدا جدا شده است که باعث مایکوتوکسیکوزهای مختلف میشود (Reeves et al., 2004). مطالعات in vitro و in vivo نشان داد که گلیوتوکسین آستروسیت های CNS را در مدل های موش مورد هدف قرار می دهد (Willis et al., 2004). علاوه بر این، CSF درمان شده با حرارت به دست آمده از بیماران ام اس باعث مرگ آپوپتوز آستروسیت ها و الیگودندروسیت ها می شود (Purzycki and Shain، ۲۰۱۰). قابل توجه است که CSF از بیماران مبتلا به سایر اختلالات عصبی التهابی یا غیر التهابی هیچ سمیتی علیه این سلول ها نشان نداد. مطالعه دیگری نشان داده است که تزریق داخل بطنی CSF حاوی گلیوتوکسین باعث افزایش نفوذپذیری سد خونی مغزی می شود که منجر به نشت ایمونوگلوبولین ها می شود (Rieger et al., 1996).
نتیجه
برای چندین دهه، عوامل ایجاد کننده اختلالات عصبی ناشناخته بودند. به طور کلی پذیرفته شده است که بسیاری از اختلالات تخریب عصبی نتیجه جهش های ژنتیکی، التهاب، پروتئین های نادرست چین خورده و عوامل محیطی هستند. اگرچه این درست است، اما سهم سایر بیماری ها را در نظر نمی گیرد. فرض دیگر ارتباط عفونت های میکروبی و علت بیماری های تخریب عصبی است. این فرض حتی به طور منطقی ارتباط مستقیمی با سن ایجاد می کند، زیرا افراد با سنین بالاتر در معرض تعداد بیشتری از بیماری های میکروبی هستند و همچنین خطر بیشتری برای آشکار شدن اختلالات تخریب عصبی دارند. میکروارگانیسم ها می توانند بر رشد مغز، سطوح انتقال دهنده های عصبی، سطح فاکتور نوتروفیلی مشتق از مغز و سطح آمیگدال تأثیر بگذارند. به محض عفونت، میکروارگانیسمها میتوانند باعث آزاد شدن پروتئینهای مختلف سیگنالدهنده سلولی شوند که باعث التهاب میشوند. هنگامی که این میکروارگانیسم ها از سد خونی مغزی عبور می کنند، این التهاب معمولاً با انحطاط نورون برگشت ناپذیر خاتمه می یابد و علائم مشابهی با اختلالات تخریب عصبی مختلف ایجاد می کند. تأثیر درمان بیماریهای میکروبی بر بیماریهای تخریبکننده عصبی، زمینهای جالب برای کار آینده است و ممکن است حداقل شروع این بیماریها را به تعویق بیندازد. علاوه بر این، بروز بالقوه اختلالات نورودژنراتیو در هر جمعیت معین ممکن است از طریق آسیب پذیری سیستم ایمنی بدن در برابر بیماری های تخریب عصبی مرتبط با بیماری های میکروبی تعیین شود. علاوه بر این، سابقه سلامتی هر فرد مبتلا به بیماریهای تخریب عصبی مرتبط با بیماریهای میکروبی ممکن است حساسیت آنها را به اختلالات تخریب عصبی توضیح دهد.
مشارکتهای نویسنده
MD و HKSP هر دو دستنوشته را نوشتند و GG آن را ویرایش، تایید و نهایی کردند.
بیانیه تعارض منافع
نویسندگان اعلام می کنند که این تحقیق در غیاب هر گونه روابط تجاری یا مالی که می تواند به عنوان تضاد منافع بالقوه تعبیر شود، انجام شده است.
پانویسها و منابع
منابع مالی. MD و HKSP هر دو توسط یک بورس تحصیلی بین المللی از دانشگاه Macquarie پشتیبانی می شوند. پروژه تحقیقاتی GG توسط شورای تحقیقات استرالیا (ARC) تامین می شود.
اطلاعات مقاله
Article information
منابع
References
- Agnihotri S. P., Dang X., Carter J. L., Fife T. D., Bord E., Batson S., et al.. (2014). JCV GCN in a natalizumab-treated MS patient is associated with mutations of the VP1 capsid gene. Neurology ۸۳, ۷۲۷–۷۳۲. ۱۰.۱۲۱۲/wnl.0000000000000713 [PMC free article] [PubMed] [CrossRef] [Google Scholar]
- Albright A. V., González-Scarano F. (2004). Microarray analysis of activated mixed glial (microglia) and monocyte-derived macrophage gene expression. J. Neuroimmunol. ۱۵۷, ۲۷–۳۸. ۱۰.۱۰۱۶/j.jneuroim.2004.09.007 [PubMed] [CrossRef] [Google Scholar]
- Alonso R., Pisa D., Rabano A., Carrasco L. (2014). Alzheimer’s disease and disseminated mycoses. Eur. J. Clin. Microbiol. Infect. Dis. ۳۳, ۱۱۲۵–۱۱۳۲. ۱۰.۱۰۰۷/s10096-013-2045-z [PubMed] [CrossRef] [Google Scholar]
- Asti A., Gioglio L. (2014). Can a bacterial endotoxin be a key factor in the kinetics of amyloid fibril formation? J. Alzheimers Dis. ۳۹, ۱۶۹–۱۷۹. ۱۰.۳۲۳۳/jad-131394 [PubMed] [CrossRef] [Google Scholar]
- Aziz Q., Doré J., Emmanuel A., Guarner F., Quigley E. (2013). Gut microbiota and gastrointestinal health: current concepts and future directions. Neurogastroenterol. Motil. ۲۵, ۴–۱۵. ۱۰.۱۱۱۱/nmo.12046 [PubMed] [CrossRef] [Google Scholar]
- Balin B. J., Gérard H. C., Arking E. J., Appelt D. M., Branigan P. J., Abrams J. T., et al.. (1998). Identification and localization of Chlamydia pneumoniae in the Alzheimer’s brain. Med. Microbiol. Immunol. ۱۸۷, ۲۳–۴۲. ۱۰.۱۰۰۷/s004300050071 [PubMed] [CrossRef] [Google Scholar]
- Beilina A., Cookson M. R. (2016). Genes associated with Parkinson’s disease: regulation of autophagy and beyond. J. Neurochem. ۱۳۹, ۹۱–۱۰۷. ۱۰.۱۱۱۱/jnc.13266 [PMC free article] [PubMed] [CrossRef] [Google Scholar]
- Benito-León J., Pisa D., Alonso R., Calleja P., Díaz-Sánchez M., Carrasco L. (2010). Association between multiple sclerosis and Candida species: evidence from a case-control study. Eur. J. Clin. Microbiol. Infect. Dis. ۲۹, ۱۱۳۹–۱۱۴۵. ۱۰.۱۰۰۷/s10096-010-0979-y [PubMed] [CrossRef] [Google Scholar]
- Berer K., Mues M., Koutrolos M., Al Rasbi Z., Boziki M., Johner C., et al.. (2011). Commensal microbiota and myelin autoantigen cooperate to trigger autoimmune demyelination. Nature ۴۷۹, ۵۳۸–۵۴۱. ۱۰.۱۰۳۸/nature10554 [PubMed] [CrossRef] [Google Scholar]
- Bernal-Cano F., Joseph J., Koralnik I. (2007). Spinal cord lesions of progressive multifocal leukoencephalopathy in an acquired immunodeficiency syndrome patient. J. Neurovirol. ۱۳, ۴۷۴–۴۷۶. ۱۰.۱۰۸۰/۱۳۵۵۰۲۸۰۷۰۱۴۶۹۱۷۸ [PubMed] [CrossRef] [Google Scholar]
- Bhattacharjee S., Lukiw W. J. (2013). Alzheimer’s disease and the microbiome. Front. Cell. Neurosci. ۷:۱۵۳. ۱۰.۳۳۸۹/fncel.2013.00153 [PMC free article] [PubMed] [CrossRef] [Google Scholar]
- Boulanger L. M. (2009). Immune proteins in brain development and synaptic plasticity. Neuron ۶۴, ۹۳–۱۰۹. ۱۰.۱۰۱۶/j.neuron.2009.09.001 [PubMed] [CrossRef] [Google Scholar]
- Brettschneider J., Del Tredici K., Lee V. M.-Y., Trojanowski J. Q. (2015). Spreading of pathology in neurodegenerative diseases: a focus on human studies. Nat. Rev. Neurosci. ۱۶, ۱۰۹–۱۲۰. ۱۰.۱۰۳۸/nrn3887 [PMC free article] [PubMed] [CrossRef] [Google Scholar]
- Brissette C. A., Rossmann E., Bowman A., Cooley A. E., Riley S. P., Hunfeld K.-P., et al.. (2010). The borrelial fibronectin-binding protein RevA is an early antigen of human Lyme disease. Clin. Vaccine Immunol. ۱۷, ۲۷۴–۲۸۰. ۱۰.۱۱۲۸/cvi.00437-09 [PMC free article] [PubMed] [CrossRef] [Google Scholar]
- Brümmendorf T., Lemmon V. (2001). Immunoglobulin superfamily receptors: cis-interactions, intracellular adapters and alternative splicing regulate adhesion. Curr. Opin. Cell Biol. ۱۳, ۶۱۱–۶۱۸. ۱۰.۱۰۱۶/s0955-0674(00)00259-3 [PubMed] [CrossRef] [Google Scholar]
- Burdo T. H., Weiffenbach A., Woods S. P., Letendre S., Ellis R. J., Williams K. C. (2013). Elevated sCD163 in plasma but not cerebrospinal fluid is a marker of neurocognitive impairment in HIV infection. AIDS ۲۷, ۱۳۸۷–۱۳۹۵. ۱۰.۱۰۹۷/qad.0b013e32836010bd [PMC free article] [PubMed] [CrossRef] [Google Scholar]
- Cantarel B. L., Waubant E., Chehoud C., Kuczynski J., Desantis T. Z., Warrington J., et al.. (2015). Gut microbiota in multiple sclerosis: possible influence of immunomodulators. J. Investig. Med. ۶۳, ۷۲۹–۷۳۴. ۱۰.۱۰۹۷/jim.0000000000000192 [PMC free article] [PubMed] [CrossRef] [Google Scholar]
- Cavanagh J., Holton J., Nolan C., Ray D., Naik J., Mantle P. (1998). The effects of the tremorgenic mycotoxin penitrem A on the rat cerebellum. Vet. Pathol. ۳۵, ۵۳–۶۳. ۱۰.۱۱۷۷/۰۳۰۰۹۸۵۸۹۸۰۳۵۰۰۱۰۵ [PubMed] [CrossRef] [Google Scholar]
- Chen J., Chia N., Kalari K. R., Yao J. Z., Novotna M., Soldan M. M. P., et al.. (2016). Multiple sclerosis patients have a distinct gut microbiota compared to healthy controls. Sci. Rep. ۶:۲۸۴۸۴. ۱۰.۱۰۳۸/srep28484 [PMC free article] [PubMed] [CrossRef] [Google Scholar]
- Choroszy-Król I., Frej-Mądrzak M., Hober M., Sarowska J., Jama-Kmiecik A. (2014). Infections caused by Chlamydophila pneumoniae. Adv. Clin. Exp. Med. ۲۳, ۱۲۳–۱۲۶. ۱۰.۱۷۲۱۹/acem/37035 [PubMed] [CrossRef] [Google Scholar]
- Clarke G., Grenham S., Scully P., Fitzgerald P., Moloney R., Shanahan F., et al.. (2013). The microbiome-gut-brain axis during early life regulates the hippocampal serotonergic system in a sex-dependent manner. Mol. Psychiatry ۱۸, ۶۶۶–۶۷۳. ۱۰.۱۰۳۸/mp.2012.77 [PubMed] [CrossRef] [Google Scholar]
- Cohen R. A., Seider T. R., Navia B. (2015). HIV effects on age-associated neurocognitive dysfunction: premature cognitive aging or neurodegenerative disease? J. Mol. Model. ۷:۳۷. ۱۰.۱۱۸۶/s13195-015-0123-4 [PMC free article] [PubMed] [CrossRef] [Google Scholar]
- Crack P. J., Bray P. J. (2007). Toll-like receptors in the brain and their potential roles in neuropathology. Immunol. Cell Biol. ۸۵, ۴۷۶–۴۸۰. ۱۰.۱۰۳۸/sj.icb.7100103 [PubMed] [CrossRef] [Google Scholar]
- Del Tredici K., Braak H. (2008). A not entirely benign procedure: progression of Parkinson’s disease. Acta Neuropathol. ۱۱۵, ۳۷۹–۳۸۴. ۱۰.۱۰۰۷/s00401-008-0355-5 [PubMed] [CrossRef] [Google Scholar]
- Desbonnet L., Clarke G., Traplin A., O’sullivan O., Crispie F., Moloney R. D., et al.. (2015). Gut microbiota depletion from early adolescence in mice: implications for brain and behaviour. Brain Behav. Immun. ۴۸, ۱۶۵–۱۷۳. ۱۰.۱۰۱۶/j.bbi.2015.04.004 [PubMed] [CrossRef] [Google Scholar]
- Dittmar S., Harms H., Runkler N., Maisner A., Kim K. S., Schneider-Schaulies J. (2008). Measles virus-induced block of transendothelial migration of T lymphocytes and infection-mediated virus spread across endothelial cell barriers. J. Virol. ۸۲, ۱۱۲۷۳–۱۱۲۸۲. ۱۰.۱۱۲۸/jvi.00775-08 [PMC free article] [PubMed] [CrossRef] [Google Scholar]
- El-Ansary A., Al-Ayadhi L. (2014). Relative abundance of short chain and polyunsaturated fatty acids in propionic acid-induced autistic features in rat pups as potential markers in autism. Lipids Health Dis. ۱۳:۱۴۰. ۱۰.۱۱۸۶/۱۴۷۶-۵۱۱x-13-140 [PMC free article] [PubMed] [CrossRef] [Google Scholar]
- Erdman S., Poutahidis T. (2014). Probiotic ‘glow of health’: it’s more than skin deep. Benef. Microbes ۵, ۱۰۹–۱۱۹. ۱۰.۳۹۲۰/BM2013.0042 [PMC free article] [PubMed] [CrossRef] [Google Scholar]
- Erickson M. A., Banks W. A. (2013). Blood-brain barrier dysfunction as a cause and consequence of Alzheimer’s disease. J. Cereb. Blood Flow Metab. ۳۳, ۱۵۰۰–۱۵۱۳. ۱۰.۱۰۳۸/jcbfm.2013.135 [PMC free article] [PubMed] [CrossRef] [Google Scholar]
- Everall I., Heaton R., Marcotte T., Ellis R., Mccutchan J., Atkinson J., et al.. (1999). Cortical synaptic density is reduced in mild to moderate human immunodeficiency virus neurocognitive disorder. Brain Pathol. ۹, ۲۰۹–۲۱۷. ۱۰.۱۱۱۱/j.1750-3639.1999.tb00219.x [PMC free article] [PubMed] [CrossRef] [Google Scholar]
- Fallon B. A., Nields J. A. (1994). Lyme disease: a neuropsychiatric illness. Am. J. Psychiatry ۱۵۱, ۱۵۷۱–۱۵۸۳. ۱۰.۱۱۷۶/ajp.151.11.1571 [PubMed] [CrossRef] [Google Scholar]
- Foster J. A., Lyte M., Meyer E., Cryan J. F. (2016). Gut microbiota and brain function: an evolving field in neuroscience. Int. J. Neuropsychopharmacol. ۱۹:pyv114. 10.1093/ijnp/pyv114 [PMC free article] [PubMed] [CrossRef] [Google Scholar]
- Gérard H. C., Wildt K. L., Whittum-Hudson J. A., Lai Z., Ager J., Hudson A. P. (2005). The load of Chlamydia pneumoniae in the Alzheimer’s brain varies with APOE genotype. Microb. Pathog. ۳۹, ۱۹–۲۶. ۱۰.۱۰۱۶/j.micpath.2005.05.002 [PubMed] [CrossRef] [Google Scholar]
- Gil C., González A. A. S., León I. L., Rivera A., Olea R. S., Cedillo L. (2014). Detection of mycoplasmas in patients with amyotrophic lateral sclerosis. Adv. Microbiol. ۴, ۷۱۲–۷۱۹. ۱۰.۴۲۳۶/aim.2014.411077 [CrossRef] [Google Scholar]
- Gray F., Chrétien F., Vallat-Decouvelaere A. V., Scaravilli F. (2003). The changing pattern of HIV neuropathology in the HAART era. J. Neuropathol. Exp. Neurol. ۶۲, ۴۲۹–۴۴۰. ۱۰.۱۰۹۳/jnen/62.5.429 [PubMed] [CrossRef] [Google Scholar]
- Harezlak J., Buchthal S., Taylor M., Schifitto G., Zhong J., Daar E., et al.. (2011). Persistence of HIV−associated cognitive impairment, inflammation and neuronal injury in era of highly active antiretroviral treatment. AIDS ۲۵, ۶۲۵–۶۳۳. ۱۰.۱۰۹۷/QAD.0b013e3283427da7 [PMC free article] [PubMed] [CrossRef] [Google Scholar]
- Heaton R., Clifford D., Franklin D., Woods S., Ake C., Vaida F., et al.. (2010). HIV-associated neurocognitive disorders persist in the era of potent antiretroviral therapy CHARTER Study. Neurology ۷۵, ۲۰۸۷–۲۰۹۶. ۱۰.۱۲۱۲/WNL.0b013e318200d727 [PMC free article] [PubMed] [CrossRef] [Google Scholar]
- Hebert L. E., Weuve J., Scherr P. A., Evans D. A. (2013). Alzheimer disease in the United States (2010–۲۰۵۰) estimated using the 2010 census. Neurology ۸۰, ۱۷۷۸–۱۷۸۳. ۱۰.۱۲۱۲/WNL.0b013e31828726f5 [PMC free article] [PubMed] [CrossRef] [Google Scholar]
- Diaz Heijtz R., Wang S., Anuar F., Qian Y., Björkholm B., Samuelsson A., et al.. (2011). Normal gut microbiota modulates brain development and behavior. Proc. Natl. Acad. Sci. U S A ۱۰۸, ۳۰۴۷–۳۰۵۲. ۱۰.۱۰۷۳/pnas.1010529108 [PMC free article] [PubMed] [CrossRef] [Google Scholar]
- Kannarkat G. T., Boss J. M., Tansey M. G. (2013). The role of innate and adaptive immunity in Parkinson’s disease. J. Parkinsons Dis. ۳, ۴۹۳–۵۱۴. ۱۰.۳۲۳۳/JPD-130250 [PMC free article] [PubMed] [CrossRef] [Google Scholar]
- Jessen Krut J., Mellberg T., Price R. W., Hagberg L., Fuchs D., Rosengren L., et al.. (2014). Biomarker evidence of axonal injury in neuroasymptomatic HIV-1 patients. PLoS One ۹:e88591. 10.1371/journal.pone.0088591 [PMC free article] [PubMed] [CrossRef] [Google Scholar]
- Levin M. C., Lee S. M., Kalume F., Morcos Y., Dohan F. C., Jr., Hasty K. A., et al.. (2002). Autoimmunity due to molecular mimicry as a cause of neurological disease. Nat. Med. ۸, ۵۰۹–۵۱۳. ۱۰.۱۰۳۸/nm0502-509 [PMC free article] [PubMed] [CrossRef] [Google Scholar]
- Lindsey J. W., deGannes S. L., Pate K. A., Zhao X. (2016). Antibodies specific for Epstein-Barr virus nuclear antigen-1 cross-react with human heterogeneous nuclear ribonucleoprotein L. Mol. Immunol. ۶۹, ۷–۱۲. ۱۰.۱۰۱۶/j.molimm.2015.11.007 [PMC free article] [PubMed] [CrossRef] [Google Scholar]
- Lipton S. A., Hickey W. F., Morris J. H., Loscalzo J. (1984). Candidal infection in the central nervous system. Am. J. Med. ۷۶, ۱۰۱–۱۰۸. ۱۰.۱۰۱۶/۰۰۰۲-۹۳۴۳(۸۴)۹۰۷۵۷-۵ [PubMed] [CrossRef] [Google Scholar]
- Lomakin Y., Arapidi G. P., Chernov A., Ziganshin R., Tcyganov E., Lyadova I., et al.. (2017). Exposure to the Epstein-Barr Viral antigen latent Membrane Protein 1 induces Myelin-reactive antibodies in vivo. Front. Immunol. ۸:۷۷۷. ۱۰.۳۳۸۹/fimmu.2017.00777 [PMC free article] [PubMed] [CrossRef] [Google Scholar]
- Luczynski P., Whelan S. O., O’sullivan C., Clarke G., Shanahan F., Dinan T. G., et al.. (2016). Adult microbiota-deficient mice have distinct dendritic morphological changes: differential effects in the amygdala and hippocampus. Eur. J. Neurosci. ۴۴, ۲۶۵۴–۲۶۶۶. ۱۰.۱۱۱۱/ejn.13291 [PMC free article] [PubMed] [CrossRef] [Google Scholar]
- Lyons J. L., Uno H., Ancuta P., Kamat A., Moore D. J., Singer E. J., et al.. (2011). Plasma sCD14 is a biomarker associated with impaired neurocognitive test performance in attention and learning domains in HIV infection. J. Acquir. Immune Defic. Syndr. ۵۷, ۳۷۱–۳۷۹. ۱۰.۱۰۹۷/qai.0b013e3182237e54 [PMC free article] [PubMed] [CrossRef] [Google Scholar]
- Lyte M. (2014). Microbial endocrinology: host-microbiota neuroendocrine interactions influencing brain and behavior. Gut Microbes ۵, ۳۸۱–۳۸۹. ۱۰.۴۱۶۱/gmic.28682 [PMC free article] [PubMed] [CrossRef] [Google Scholar]
- MacDonald A. B., Miranda J. M. (1987). Concurrent neocortical borreliosis and Alzheimer’s disease. Hum. Pathol. ۱۸, ۷۵۹–۷۶۱. ۱۰.۱۰۱۶/s0046-8177(87)80252-6 [PubMed] [CrossRef] [Google Scholar]
- Malmeström C., Haghighi S., Rosengren L., Andersen O., Lycke J. (2003). Neurofilament light protein and glial fibrillary acidic protein as biological markers in MS. Neurology ۶۱, ۱۷۲۰–۱۷۲۵. ۱۰.۱۲۱۲/۰۱.wnl.0000098880.19793.b6 [PubMed] [CrossRef] [Google Scholar]
- McCoy A. J., Adams N. E., Hudson A. O., Gilvarg C., Leustek T., Maurelli A. T. (2006). L,L-diaminopimelate aminotransferase, a trans-kingdom enzyme shared by Chlamydia and plants for synthesis of diaminopimelate/lysine. Proc. Natl. Acad. Sci. U S A ۱۰۳, ۱۷۹۰۹–۱۷۹۱۴. ۱۰.۱۰۷۳/pnas.0608643103 [PMC free article] [PubMed] [CrossRef] [Google Scholar]
- McGeer P. L., McGeer E. G. (2002). Local neuroinflammation and the progression of Alzheimer’s disease. J. Neurovirol. ۸, ۵۲۹–۵۳۸. ۱۰.۱۰۸۰/۱۳۵۵۰۲۸۰۲۹۰۱۰۰۹۶۹ [PubMed] [CrossRef] [Google Scholar]
- McQuaid S., Cosby S. L. (2002). An immunohistochemical study of the distribution of the measles virus receptors, CD46 and SLAM, in normal human tissues and subacute sclerosing panencephalitis. Lab. Invest. ۸۲, ۴۰۳–۴۰۹. ۱۰.۱۰۳۸/labinvest.3780434 [PubMed] [CrossRef] [Google Scholar]
- Miklossy J. (2011). Alzheimer’s disease-a neurospirochetosis. Analysis of the evidence following Koch’s and Hill’s criteria. J. Neuroinflammation ۸:۹۰. ۱۰.۱۱۸۶/۱۷۴۲-۲۰۹۴-۸-۹۰ [PMC free article] [PubMed] [CrossRef] [Google Scholar]
- Miklossy J. (2015). Historic evidence to support a causal relationship between spirochetal infections and Alzheimer’s disease. Front. Aging Neurosci. ۷:۴۶. ۱۰.۳۳۸۹/fnagi.2015.00046 [PMC free article] [PubMed] [CrossRef] [Google Scholar]
- Miklossy J., Khalili K., Gern L., Ericson R. L., Darekar P., Bolle L., et al.. (2004). Borrelia burgdorferi persists in the brain in chronic lyme neuroborreliosis and may be associated with Alzheimer disease. J. Alzheimers Dis. ۶, ۶۳۹–۶۴۹. ۱۰.۳۲۳۳/jad-2004-6608 [PubMed] [CrossRef] [Google Scholar]
- Minoretti P., Gazzaruso C., Di Vito C., Emanuele E., Bianchi M., Coen E., et al.. (2006). Effect of the functional toll-like receptor 4 Asp299Gly polymorphism on susceptibility to late-onset Alzheimer’s disease. Neurosci. Lett. ۳۹۱, ۱۴۷–۱۴۹. ۱۰.۱۰۱۶/j.neulet.2005.08.047 [PubMed] [CrossRef] [Google Scholar]
- Miskin D. P., Koralnik I. J. (2015). Novel syndromes associated with JC virus infection of neurons and meningeal cells: no longer a gray area. Curr. Opin. Neurol. ۲۸, ۲۸۸–۲۹۴. ۱۰.۱۰۹۷/wco.0000000000000201 [PMC free article] [PubMed] [CrossRef] [Google Scholar]
- Moll N., Rietsch A., Ransohoff A., Cossoy M., Huang D., Eichler F., et al.. (2008). Cortical demyelination in PML and MS Similarities and differences. Neurology ۷۰, ۳۳۶–۳۴۳. ۱۰.۱۲۱۲/۰۱.WNL.0000284601.54436.e4 [PubMed] [CrossRef] [Google Scholar]
- Najafipoor A., Roghanian R., Zarkesh-Esfahani S. H., Bouzari M., Etemadifar M. (2015). The beneficial effects of vitamin D3 on reducing antibody titers against Epstein-Barr virus in multiple sclerosis patients. Cell Immunol. ۲۹۴, ۹–۱۲. ۱۰.۱۰۱۶/j.cellimm.2015.01.009 [PubMed] [CrossRef] [Google Scholar]
- Neufeld K., Kang N., Bienenstock J., Foster J. (2011). Reduced anxiety-like behavior and central neurochemical change in germ-free mice. Neurogastroenterol. Motil. ۲۳, ۲۵۵–۲۶۴. ۱۰.۱۱۱۱/j.1365-2982.2010.01620.x [PubMed] [CrossRef] [Google Scholar]
- Nicolson G. L., Nasralla M. Y., Haier J., Pomfret J. (2002). High frequency of systemic mycoplasmal infections in Gulf War veterans and civilians with Amyotrophic Lateral Sclerosis (ALS). J. Clin. Neurosci. ۹, ۵۲۵–۵۲۹. ۱۰.۱۰۵۴/jocn.2001.1075 [PubMed] [CrossRef] [Google Scholar]
- Noguchi H., Moore J. W. (1913). A demonstration of Treponema pallidum in the brain in cases of general paralysis. J. Exp. Med. ۱۷, ۲۳۲–۲۳۸. ۱۰.۱۰۸۴/jem.17.2.232 [PMC free article] [PubMed] [CrossRef] [Google Scholar]
- Ogbonnaya E. S., Clarke G., Shanahan F., Dinan T. G., Cryan J. F., O’leary O. F. (2015). Adult hippocampal neurogenesis is regulated by the microbiome. Biol. Psychiatry ۷۸, e7–e9. 10.1016/j.biopsych.2014.12.023 [PubMed] [CrossRef] [Google Scholar]
- Paradowski B., Jaremko M., Dobosz T., Leszek J., Noga L. (2007). Evaluation of CSF-Chlamydia pneumoniae, CSF-tau, and CSF-Aβ۴۲ in Alzheimer’s disease and vascular dementia. J. Neurol. ۲۵۴, ۱۵۴–۱۵۹. ۱۰.۱۰۰۷/s00415-006-0298-5 [PubMed] [CrossRef] [Google Scholar]
- Pasol J., Feuer W., Yang C., Shaw G., Kardon R., Guy J. (2010). Phosphorylated neurofilament heavy chain correlations to visual function, optical coherence tomography, and treatment. Mult. Scler. Int. ۲۰۱۰:۵۴۲۶۹۱. ۱۰.۱۱۵۵/۲۰۱۰/۵۴۲۶۹۱ [PMC free article] [PubMed] [CrossRef] [Google Scholar]
- Patterson J. B., Cornu T. I., Redwine J., Dales S., Lewicki H., Holz A., et al.. (2001). Evidence that the hypermutated M protein of a subacute sclerosing panencephalitis measles virus actively contributes to the chronic progressive CNS disease. Virology ۲۹۱, ۲۱۵–۲۲۵. ۱۰.۱۰۰۶/viro.2001.1182 [PubMed] [CrossRef] [Google Scholar]
- Pisa D., Alonso R., Juarranz A., Rábano A., Carrasco L. (2015). Direct visualization of fungal infection in brains from patients with Alzheimer’s disease. J. Alzheimers Dis. ۴۳, ۶۱۳–۶۲۴. ۱۰.۳۲۳۳/jad-141386 [PubMed] [CrossRef] [Google Scholar]
- Poulsen D. J., Robertson S. J., Favara C. A., Portis J. L., Chesebro B. W. (1998). Mapping of a neurovirulence determinant within the envelope protein of a polytropic murine retrovirus: induction of central nervous system disease by low levels of virus. Virology ۲۴۸, ۱۹۹–۲۰۷. ۱۰.۱۰۰۶/viro.1998.9258 [PubMed] [CrossRef] [Google Scholar]
- Power C. (2001). Retroviral diseases of the nervous system: pathogenic host response or viral gene-mediated neurovirulence? Trends Neurosci. ۲۴, ۱۶۲–۱۶۹. ۱۰.۱۰۱۶/s0166-2236(00)01737-9 [PubMed] [CrossRef] [Google Scholar]
- Prusiner S. B., Woerman A. L., Mordes D. A., Watts J. C., Rampersaud R., Berry D. B., et al.. (2015). Evidence for α-synuclein prions causing multiple system atrophy in humans with parkinsonism. Proc. Natl. Acad. Sci. U S A ۱۱۲, E5308–E5317. 10.1073/pnas.1514475112 [PMC free article] [PubMed] [CrossRef] [Google Scholar]
- Purzycki C. B., Shain D. H. (2010). Fungal toxins and multiple sclerosis: a compelling connection. Brain Res. Bull. ۸۲, ۴–۶. ۱۰.۱۰۱۶/j.brainresbull.2010.02.012 [PubMed] [CrossRef] [Google Scholar]
- Reeves E. P., Messina C. G. M., Doyle S., Kavanagh K. (2004). Correlation between gliotoxin production and virulence of Aspergillus fumigatus in Galleria mellonella. Mycopathologia ۱۵۸, ۷۳–۷۹. ۱۰.۱۰۲۳/B:MYCO.0000038434.55764.16 [PubMed] [CrossRef] [Google Scholar]
- Reuter D., Schneider-Schaulies J. (2010). Measles virus infection of the CNS: human disease, animal models, and approaches to therapy. Med. Microbiol. Immunol. ۱۹۹, ۲۶۱–۲۷۱. ۱۰.۱۰۰۷/s00430-010-0153-2 [PubMed] [CrossRef] [Google Scholar]
- Rieger F., Amouri R., Benjelloun N., Cifuentes-Diaz C., Lyon-Caen O., Hantaz-Ambroise D., et al.. (1996). Gliotoxic factor and multiple sclerosis. C. R. Acad. Sci. III ۳۱۹, ۳۴۳–۳۵۰. [PubMed] [Google Scholar]
- Riviere G. R., Riviere K., Smith K. (2002). Molecular and immunological evidence of oral Treponema in the human brain and their association with Alzheimer’s disease. Oral Microbiol. Immunol. ۱۷, ۱۱۳–۱۱۸. ۱۰.۱۰۴۶/j.0902-0055.2001.00100.x [PubMed] [CrossRef] [Google Scholar]
- Sampson T. R., Debelius J. W., Thron T., Janssen S., Shastri G. G., Ilhan Z. E., et al.. (2016). Gut microbiota regulate motor deficits and neuroinflammation in a model of Parkinson’s disease. Cell ۱۶۷, ۱۴۶۹.e12–۱۴۸۰.e12. 10.1016/j.cell.2016.11.018 [PMC free article] [PubMed] [CrossRef] [Google Scholar]
- Santiago O., Gutierrez J., Sorlozano A., de Dios Luna J., Villegas E., Fernandez O. (2010). Relation between Epstein-Barr virus and multiple sclerosis: analytic study of scientific production. Eur. J. Clin. Microbiol. Infect. Dis. ۲۹, ۸۵۷–۸۶۶. ۱۰.۱۰۰۷/s10096-010-0940-0 [PubMed] [CrossRef] [Google Scholar]
- Sei Y., Kustova Y., Li Y., Morse H. C., III., Skolnick P., Basile A. S. (1998). The encephalopathy associated with murine acquired immunodeficiency syndrome. Ann. N Y Acad. Sci. ۸۴۰, ۸۲۲–۸۳۴. ۱۰.۱۱۱۱/j.1749-6632.1998.tb09620.x [PubMed] [CrossRef] [Google Scholar]
- Serafini B., Rosicarelli B., Franciotta D., Magliozzi R., Reynolds R., Cinque P., et al.. (2007). Dysregulated Epstein-Barr virus infection in the multiple sclerosis brain. J. Exp. Med. ۲۰۴, ۲۸۹۹–۲۹۱۲. ۱۰.۱۰۸۴/jem.20071030 [PMC free article] [PubMed] [CrossRef] [Google Scholar]
- Sriram S., Stratton C. W., Yao S. Y., Tharp A., Ding L., Bannan J. D., et al.. (1999). Chlamydia pneumoniae infection of the central nervous system in multiple sclerosis. Ann. Neurol. ۴۶, ۶–۱۴. ۱۰.۱۰۰۲/۱۵۳۱-۸۲۴۹(۱۹۹۹۰۷)۴۶:۱<6::aid-ana4>3.3.co;2-d [PubMed] [CrossRef] [Google Scholar]
- Stilling R. M., Ryan F. J., Hoban A. E., Shanahan F., Clarke G., Claesson M. J., et al.. (2015). Microbes and neurodevelopment—absence of microbiota during early life increases activity-related transcriptional pathways in the amygdala. Brain Behav. Immun. ۵۰, ۲۰۹–۲۲۰. ۱۰.۱۰۱۶/j.bbi.2015.07.009 [PubMed] [CrossRef] [Google Scholar]
- Stilling R. M., van de Wouw M., Clarke G., Stanton C., Dinan T. G., Cryan J. F. (2016). The neuropharmacology of butyrate: the bread and butter of the microbiota-gut-brain axis? Neurochem. Int. ۹۹, ۱۱۰–۱۳۲. ۱۰.۱۰۱۶/j.neuint.2016.06.011 [PubMed] [CrossRef] [Google Scholar]
- Stockmann-Juvala H., Savolainen K. (2008). A review of the toxic effects and mechanisms of action of fumonisin B1. Hum. Exp. Toxicol. ۲۷, ۷۹۹–۸۰۹. ۱۰.۱۱۷۷/۰۹۶۰۳۲۷۱۰۸۰۹۹۵۲۵ [PubMed] [CrossRef] [Google Scholar]
- Tahara K., Kim H.-D., Jin J.-J., Maxwell J. A., Li L., Fukuchi K.-I. (2006). Role of toll-like receptor signalling in Aβ uptake and clearance. Brain ۱۲۹, ۳۰۰۶–۳۰۱۹. ۱۰.۱۰۹۳/brain/awl249 [PMC free article] [PubMed] [CrossRef] [Google Scholar]
- Tomova A., Husarova V., Lakatosova S., Bakos J., Vlkova B., Babinska K., et al.. (2015). Gastrointestinal microbiota in children with autism in Slovakia. Physiol. Behav. ۱۳۸, ۱۷۹–۱۸۷. ۱۰.۱۰۱۶/j.physbeh.2014.10.033 [PubMed] [CrossRef] [Google Scholar]
- Torkildsen Ø., Stansberg C., Angelskår S. M., Kooi E. J., Geurts J. J., van der Valk P., et al.. (2010). Upregulation of immunoglobulin-related genes in cortical sections from multiple sclerosis patients. Brain Pathol. ۲۰, ۷۲۰–۷۲۹. ۱۰.۱۱۱۱/j.1750-3639.2009.00343.x [PMC free article] [PubMed] [CrossRef] [Google Scholar]
- Turkel Y., Dag E., Gunes H., Apan T., Yoldas T. (2015). Is there a relationship between Parkinson’s disease and Chlamydia pneumoniae? Niger. J. Clin. Pract. ۱۸, ۶۱۲–۶۱۵. ۱۰.۴۱۰۳/۱۱۱۹-۳۰۷۷.۱۵۴۲۱۵ [PubMed] [CrossRef] [Google Scholar]
- Valcour V., Chalermchai T., Sailasuta N., Marovich M., Lerdlum S., Suttichom D., et al.. (2012). Central nervous system viral invasion and inflammation during acute HIV infection. J. Infect. Dis. ۲۰۶, ۲۷۵–۲۸۲. ۱۰.۱۰۹۳/infdis/jis326 [PMC free article] [PubMed] [CrossRef] [Google Scholar]
- Vardhini D., Suneetha S., Ahmed N., Joshi D., Karuna S., Magee X., et al.. (2004). Comparative proteomics of the Mycobacterium leprae binding protein myelin P0: its implication in leprosy and other neurodegenerative diseases. Infect. Genet. Evol. ۴, ۲۱–۲۸. ۱۰.۱۰۱۶/j.meegid.2003.11.001 [PubMed] [CrossRef] [Google Scholar]
- Viala J. P., Mochegova S. N., Meyer-Morse N., Portnoy D. A. (2008). A bacterial pore-forming toxin forms aggregates in cells that resemble those associated with neurodegenerative diseases. Cell. Microbiol. ۱۰, ۹۸۵–۹۹۳. ۱۰.۱۱۱۱/j.1462-5822.2007.01100.x [PubMed] [CrossRef] [Google Scholar]
- von Giesen H.-J., Antke C., Hefter H., Wenserski F., Seitz R. J., Arendt G. (2000). Potential time course of human immunodeficiency virus type 1-associated minor motor deficits: electrophysiologic and positron emission tomography findings. Arch. Neurol. ۵۷, ۱۶۰۱–۱۶۰۷. ۱۰.۱۰۰۱/archneur.57.11.1601 [PubMed] [CrossRef] [Google Scholar]
- Watanabe S., Shirogane Y., Sato Y., Hashiguchi T., Yanagi Y. (2018). New insights into measles virus brain infections. Trends Microbiol. [Epub ahead of print]. 10.1016/j.tim.2018.08.010 [PubMed] [CrossRef] [Google Scholar]
- Wijburg M. T., van Oosten B. W., Murk J.-L., Karimi O., Killestein J., Wattjes M. P. (2015). Heterogeneous imaging characteristics of JC virus granule cell neuronopathy (GCN): a case series and review of the literature. J. Neurol. ۲۶۲, ۶۵–۷۳. ۱۰.۱۰۰۷/s00415-014-7530-5 [PubMed] [CrossRef] [Google Scholar]
- Willis C. L., Leach L., Clarke G. J., Nolan C. C., Ray D. E. (2004). Reversible disruption of tight junction complexes in the rat blood-brain barrier, following transitory focal astrocyte loss. Glia ۴۸, ۱–۱۳. ۱۰.۱۰۰۲/glia.20049 [PubMed] [CrossRef] [Google Scholar]
- Willis S. N., Stadelmann C., Rodig S. J., Caron T., Gattenloehner S., Mallozzi S. S., et al.. (2009). Epstein-Barr virus infection is not a characteristic feature of multiple sclerosis brain. Brain ۱۳۲, ۳۳۱۸–۳۳۲۸. ۱۰.۱۰۹۳/brain/awp200 [PMC free article] [PubMed] [CrossRef] [Google Scholar]
- Wozniak M., Mee A., Itzhaki R. (2009). Herpes simplex virus type 1 DNA is located within Alzheimer’s disease amyloid plaques. J. Pathol. ۲۱۷, ۱۳۱–۱۳۸. ۱۰.۱۰۰۲/path.2449 [PubMed] [CrossRef] [Google Scholar]
- Wunderink R. G., Waterer G. W. (2014). Community-acquired pneumonia. N. Engl. J. Med. ۳۷۰, ۵۴۳–۵۵۱. ۱۰.۱۰۵۶/NEJMcp1214869 [PubMed] [CrossRef] [Google Scholar]
- Zhao Y., Lukiw W. J. (2015). Microbiome-generated amyloid and potential impact on amyloidogenesis in Alzheimer’s disease (AD). J. Nat. Sci. ۱:e138. Available online at: http://www.jnsci.org/index.php?journal=nsci&page=article&op=view&path%5B%5D=138 [PMC free article] [PubMed] [Google Scholar]
- Zhu J., Pelidou S.-H., Deretzi G., Levi M., Mix E., van der Meide P., et al.. (2001). P0 glycoprotein peptides 56–۷۱ and 180–۱۹۹ dose-dependently induce acute and chronic experimental autoimmune neuritis in Lewis rats associated with epitope spreading. J. Neuroimmunol. ۱۱۴, ۹۹–۱۰۶. ۱۰.۱۰۱۶/s0165-5728(01)00245-4 [PubMed] [CrossRef] [Google Scholar]