نقش تعیینکننده میکروبیوتای روده در بازنویسی اپیژنوم و آشکارسازی اهداف درمانی نوین در بیماریهای آلزایمر و پارکینسون

نقش تعیینکننده میکروبیوتای روده (Gut Microbiota) در بازنویسی اپیژنوم (Epigenomes) و آشکارسازی اهداف درمانی (Therapy) نوین در بیماریهای آلزایمر (Alzheimer’s Disease) و پارکینسون (Parkinson’s Disease)
چکیده (Abstract)
ریشههای بیماری آلزایمر (Alzheimer’s Disease – AD) و بیماری پارکینسون (Parkinson’s Disease – PD) شامل جهشهای ژنتیکی (Genetic Mutations)، تغییرات اپیژنتیکی (Epigenetic Changes)، قرارگیری در معرض نوروتوکسینها (Neurotoxin Exposure) و اختلال در میکروبیوتای روده (Gut Microbiota Dysregulation) است. ترکیب پویا و متغیر میکروبیوتای روده و متابولیتهای آن بر یکپارچگی سد رودهای و سد خونی–مغزی (Intestinal and Blood–Brain Barrier Integrity) تأثیر میگذارد و در توسعه AD و PD نقش دارد. این مرور علمی (Review) به بررسی تاخوردگی و تجمع پروتئینها (Protein Misfolding and Aggregation) و ارتباطات اپیژنتیکی (Epigenetic Links) در پاتوژنز AD و PD میپردازد. همچنین، نقش رودهی نفوذپذیر (Leaky Gut) و محور روده–میکروبیوتا–مغز (Microbiota–Gut–Brain Axis) در تشدید این بیماریها از طریق تغییرات اپیژنتیکی القاشده توسط التهاب (Inflammation-Induced Epigenetic Alterations) برجسته میشود. علاوه بر این، پتانسیل رژیم غذایی (Diet)، پروبیوتیکها (Probiotics) و پیوند میکروبیوتا (Microbiota Transplantation) برای پیشگیری و درمان AD و PD از طریق اصلاحات اپیژنتیکی مورد بررسی قرار گرفته و چالشهای کنونی و ملاحظات آینده (Current Challenges and Future Considerations) نیز مورد بحث قرار میگیرند. این رویکردها وعدهی قابل توجهی برای تبدیل یافتههای پژوهشی به کاربردهای بالینی عملی (Practical Clinical Applications) ارائه میدهند.
کلیدواژهها (Keywords): بیماری آلزایمر (Alzheimer’s Disease – AD)، اپیژنتیک (Epigenetic)، روده نفوذپذیر (Leaky Gut)، میکروبیوتا (Microbiota)، بیماری پارکینسون (Parkinson’s Disease – PD)، پروبیوتیکها (Probiotics)
خلاصه به زبان ساده (Plain language summary)
بیماری آلزایمر (Alzheimer’s disease – AD) و بیماری پارکینسون (Parkinson’s disease – PD) دو بیماری شایع مغزی مرتبط با افزایش سن هستند. شیوع AD تقریباً ۲۰٪ در افراد بالای ۸۰ سال و شیوع PD بین ۱ تا ۴٪ در افراد بالای ۶۰ سال است. پژوهشگران در حال بررسی روابط متنوع میان عوامل کلیدی دخیل در پاتوژنز (pathogenesis) این دو بیماری هستند، از جمله رژیم غذایی (diet)، میکروبیوتای روده (gut microbiota)، نورواکسایش (neuroinflammation)، تغییرات اپیژنتیک (epigenetic modifications) و تغییرات ژنتیکی (genetic changes) تا بینشی عمیقتر از مکانیزمهای توسعه بیماری به دست آورند و درمانهای مؤثرتری برای این بیماریهای ناتوانکننده طراحی کنند. کشف این روابط فرصتهایی برای حفظ سلامت مطلوب از طریق تغییرات رژیم غذایی–میکروبیوتا–اپیژنتیک (diet–microbiota–epigenetic modifications) فراهم میکند، زیرا رژیم غذایی (diet) و محیط اطراف نقش حیاتی در تغییرات میکروبی روده (gut microbial alterations) ایفا میکنند. در این مقاله، ما تعاملات بین تاشدگی/تجمع پروتئین مخرب (destructive protein misfolding/aggregation) در AD و PD را همراه با التهاب عصبی (neuroinflammation) و تغییرات اپیژنتیک (epigenetic alterations) بررسی میکنیم که همه این فرآیندها تحت تأثیر تغذیه (nutrition)، عدم تعادل میکروبیوتا (microbiota dysbiosis)، نشت روده – اختلال در سد روده-خون (leaky gut – gut–blood barrier disruption) و سموم داخلی یا محیطی (internal or environmental toxins) قرار دارند. همچنین بحثها و ایدههای تحریککننده تفکر درباره رویکردهای پیشگیرانه و درمانی اخیر مانند رژیمهای خاص (special diets)، پروبیوتیکها (probiotics)، پیوند میکروبیوتای مدفوعی (fecal microbiota transplantation) و حتی برخی آنتیبیوتیکهای خاص (specific antibiotics) برای پیشگیری یا بهبود علائم عصبروانپزشکی (neuropsychiatric symptoms) در AD و PD ارائه شده است.
چکیدهای مناسب برای توییت (Tweetable Abstract):
بیماریهای آلزایمر (Alzheimer’s disease – AD) و پارکینسون (Parkinson’s disease – PD) ناشی از تغییرات ژنتیکی (genetic)، اپیژنتیک (epigenetic)، تغییرات میکروبیوتای روده (gut microbiota shifts) و سموم (toxins) هستند. دیسبیوزیس میکروبیوتا (microbiota dysbiosis)، سدهای روده و مغز (gut and brain barriers) را تحت تأثیر قرار داده و باعث افزایش التهاب (inflammation)، تغییرات اپیژنتیک (epigenetic changes) و تاشدگی پروتئین (protein misfolding) میشود. رژیم غذایی (diet)، پروبیوتیکها (probiotics) و انتقال میکروبیوتای مدفوع (fecal transfer) نویدبخش مسیرهای درمانی امیدوارکننده هستند.
اختلالات نورودژنراتیو (Neurodegenerative disorders) مانند بیماری آلزایمر (Alzheimer’s disease – AD) و بیماری پارکینسون (Parkinson’s disease – PD) با از دست رفتن عملکرد نورونها (loss of neuronal functions) و اختلالات عصبی (impairment) مشخص میشوند که منجر به کاهشهای پیشرونده شناختی (progressive cognitive deficits) میشود [1]. ویژگی اصلی (core hallmark) در AD و PD، تجمع و تاشدگی پروتئینهای خاص (aggregation and misfolding of specific proteins) در داخل یا خارج سلولهای عصبی (neuronal cells) است [2]. توسعه و پیشرفت AD و PD میتواند توسط جهشهای ژنتیکی (genetic mutations) و همچنین تغییرات متنوع اپیژنتیک (epigenetic alterations) شامل miRNAها، متیلاسیون DNA (DNA methylation) و تغییرات هیستونی (histone modifications) هدایت شود [3,4]. در حالی که ۵-۱٪ موارد AD ناشی از جهشهای اتوزومال (autosomal mutations) هستند و موارد اسپادیک ۹۵٪ بیماران AD را تشکیل میدهند [5]، و در PD، ۱۶–۳۶٪ از ریسک بیماری میتواند به جهشهای ژنتیکی (genetic mutations) نسبت داده شود [6]، سامانههای اپیژنتیک (epigenetic machinery) یکی از تنظیمکنندههای اصلی ژنها و مسیرهای کلیدی مرتبط با پاتوژنز (pathogenesis) AD و PD است. این موارد شامل رسوب آمیلوئید β (amyloid β – Aβ deposition)، فسفریلاسیون بیش از حد تاو (tau hyperphosphorylation)، تولید بیش از حد و تجمع پروتئین α-سینوکلئین (α-synuclein protein aggregation)، تخریب سیناپسی و نورونی (synaptic and neuronal degeneration)، التهاب عصبی (neuroinflammation) و استرس اکسیداتیو (oxidative stress) و دیگر عوامل میشوند [7–9]. همچنین مشخص شده است که علاوه بر عوامل ژنتیکی و اپیژنتیک (genetic and epigenetic factors) دخیل در اختلالات نورودژنراتیو (neurodegenerative diseases)، میکروبیوتای روده (gut microbiota – microbiome) نقشهای مهمی در پاتوژنز بیماری (disease pathogenesis) ایفا میکند [10]. میکروبیوتای روده (gut microbiota) شامل انواع میکروارگانیسمها مانند باکتریها، ویروسها، قارچها و پروتوزوآها (bacteria, viruses, fungi, and protozoa) است که ساکنان دائمی روده (gastrointestinal – GI tract) هستند [11]. تخمین زده میشود که تقریباً ۱۰۰ تریلیون میکروب رودهای (100 trillion gut microbes) در روده وجود دارد، که ۱۰–۱/۵ برابر تعداد سلولهای بدن انسان است. ترکیب ژنتیکی کلی (overall genetic composition) میکروبیوتای روده بیش از ژنوم انسان (human genome) بوده و شامل بیش از صد برابر ژنهای متمایز نسبت به ژنوم انسان [12] و تقریباً ۲۰۰ میلیون توالی پروتئینی مختلف (200 million different protein sequences) است [13]. میکروبیوتای روده (gut microbiota) از نظر تنوع میان جمعیتهای مختلف بسیار گسترده است و تاثیر عظیمی بر سلامت یا وضعیت بیماری انسان (human health or disease state) دارد [14,15]. به عنوان مثال، نشان داده شده است که دیسبیوزیس روده (gut dysbiosis) نقش کلیدی در تخریب سد رودهای (intestinal barrier destruction) و افزایش نفوذپذیری روده (intestinal permeability) دارد [16]. افزایش نفوذپذیری روده باعث افزایش واسطههای التهابی (inflammatory mediators) مانند سیتوکینها و متابولیتهای باکتریایی (cytokines and bacterial metabolites) در گردش خون میشود، که به نوبه خود یکپارچگی سد خونی–مغزی (blood–brain barrier – BBB) را مختل کرده و موجب استرس اکسیداتیو (oxidative stress) و التهاب عصبی (neuroinflammation) میشود، که از عوامل کلیدی (key players) در توسعه و پیشرفت AD و PD هستند [17,18].
شواهد نوظهور نشان دادهاند که تغییرات میکروبیوتای رودهای (gut microbiota alterations) منجر به اختلالات اپیژنتیکی (epigenetic dysregulations) میشوند که مسیرهای متعددی همچون پروتئین آمیلوئید (amyloid protein)، استرس اکسیداتیو (oxidative stress) و التهاب عصبی (neuroinflammation) را تحت تأثیر قرار میدهند [19]. بنابراین، تعدیلات اپیژنتیکی (epigenetic modulations) القاشده توسط میکروبیوم رودهای (gut microbiome) و متابولیتهای مشتق از آن (gut microbiome-derived metabolites) میتوانند به عنوان رویکردهای درمانی اصلی برای بیماری آلزایمر (AD) و بیماری پارکینسون (PD) مطرح شوند.
در این مقاله، بهروزرسانیهایی درباره موضوعات زیر ارائه میشود:
تجمع پروتئینها (protein aggregation) در آلزایمر و پارکینسون و ارتباط آن با تغییرات اپیژنتیکی؛
نقش افزایش نفوذپذیری رودهای (elevated intestinal permeability) و پدیده روده نشتپذیر (leaky gut) در پاتوژنز (pathogenesis) آلزایمر و پارکینسون از طریق تغییرات اپیژنتیکی؛
نقش محور میکروبیوم–روده–مغز (microbiome–gut–brain axis) در بروز یا پیشگیری از آلزایمر و پارکینسون از طریق سازوکارهای اپیژنتیکی (epigenetic mechanisms)؛
و در نهایت، درمانهای هدفمند مبتنی بر میکروبیوم (microbiome-targeted therapies) به عنوان راهبردهای نوین درمانی برای آلزایمر و پارکینسون از طریق تعدیل اپیژنتیکی (epigenetic modulations).
آلزایمر (AD)، تجمع پروتئینی و ارتباط آن با تغییرات اپیژنتیکی (epigenetic changes)
بیماری آلزایمر (Alzheimer’s disease; AD) به عنوان علت اصلی زوال عقل (dementia) در افراد بالای ۶۵ سال شناخته میشود و با اختلالات پیشرونده حافظه و شناختی (progressive memory and cognitive deficits) همراه با نابهنجاریهای رفتاری (behavioral aberrations) همراه است [20]. آلزایمر یکی از اختلالات مرتبط با افزایش سن (age-related disorders) با رشد سریع است که در نتیجه برهمکنشهای پیچیده عوامل ژنتیکی، اپیژنتیکی، محیطی و سبک زندگی (genetic, epigenetic, and environmental factors as well as lifestyle) ایجاد میشود [21,22]. بهترین شاخصهای افت شناختی (cognitive decline) در آلزایمر عبارتاند از رسوب خارجسلولی آمیلوئید بتا (Aβ) به صورت پلاکهای نوریتی (neuritic plaques) و تجمع درونسلولی درهمتنیدگیهای نوروفیبریلی (neurofibrillary tangles) که ناشی از فسفریلاسیون بیشازحد پروتئین تاو (hyperphosphorylated tau) هستند [23,24]. هر دو آنزیم بتا-سکرتاز (β-secretase) و گاما-سکرتاز (γ-secretase) به طور عمده تولید آمیلوئید بتا (Aβ production) را از پروتئین پیشساز آمیلوئید (APP; amyloid precursor protein) درون اندوزومها (endosomes) میانجیگری میکنند [25]. تولید بیش از حد Aβ ناشی از جهشهای ژنی در PSEN1/2 و APP مسئول شکل اتوزومال غالب آلزایمر زودرس (autosomal dominant early-onset AD) شناخته شده است [26]. تجمع آمیلوئید بتا (Aβ aggregation) در قالب الیگومرها (oligomers) و فیبریلهای مرتبه بالاتر (higher-order fibrils) موجب فسفریلاسیون بیشازحد تاو (tau hyperphosphorylation) شده و در نتیجه جریان خون مویرگی مغزی (cerebral capillary blood flow) و فعالیت سیناپسی (synaptic activity) را مختل میکند [23]. به طور کلی، تجمع آمیلوئید بتا (Aβ aggregation) به عنوان یک فرآیند پاتولوژیک کلیدی (crucial pathological process) در برانگیختن تجمع تاو (tau accumulation) و التهاب عصبی (neuroinflammation) در نظر گرفته میشود که به نوبه خود موجب تسریع نورودژنراسیون (neurodegeneration) میگردد (شکل 1).

شکل 1. زنجیره رویدادهایی که به پاتوژنز (pathogenesis) بیماری آلزایمر (Alzheimer’s disease; AD) و بیماری پارکینسون (Parkinson’s disease; PD) منجر میشوند.
در بخش بالایی شکل، مربوط به بیماری آلزایمر (AD)، پروتئین پیشساز آمیلوئید بتا (Aβ precursor protein; APP) توسط آنزیمهای بتا-سکرتاز (β-secretase) و گاما-سکرتاز (γ-secretase) در سلولهای مغزی برش داده میشود. تجمع و انباشت آمیلوئید بتا (Aβ accumulation and aggregation) در فضای خارجسلولی موجب فعالسازی میکروگلیا (microglia activation)، همچنین فسفریلاسیون و تجمع پروتئین تاو (tau phosphorylation and aggregation) در قالب درهمتنیدگیهای نوروفیبریلی (neurofibrillary tangles) در نورونها میگردد که خود باعث تشدید فعالسازی میکروگلیا میشود. فعالسازی مزمن میکروگلیا (chronic microglial activation) با التهاب عصبی (neuroinflammation) و نورودژنراسیون (neurodegeneration) در آلزایمر ارتباط دارد. با این حال، فعالیت بدنی (physical exercise) موجب تولید هورمون ایریسین (irisin) میشود که بر گیرندههای آستروسیتی (astrocytic receptors) خود یعنی اینتگرین αV/β5 (integrin αV/β5) اثر میگذارد و از طریق کاهش مسیر پیامرسانی ERK–STAT3 (downregulation of ERK–STAT3 signaling)، آزادسازی آنزیم نپریلیزین (neprilysin) ــ که تجزیهکننده آمیلوئید بتا (Aβ-degrading enzyme) است ــ را افزایش میدهد و در نتیجه موجب پاکسازی آمیلوئید بتا (Aβ clearance) میشود. در بخش پایینی شکل، مربوط به بیماری پارکینسون (PD) در ساقه مغز (brain stem)، تجمع گسترده آلفا-سینوکلئین (α-synuclein aggregation) به شکل اجسام لوی (Lewy bodies) در مغز میانی (midbrain) رخ میدهد که منجر به التهاب (inflammation) و از بین رفتن نورونهای دوپامینرژیک (loss of dopaminergic neurons) در بخش متراکم جسم سیاه (substantia nigra pars compacta) میگردد. در همین حال، تجمعات آلفا-سینوکلئین میتوانند به دیگر نواحی مغز گسترش یابند و افزون بر اختلالات حرکتی (motor dysfunction)، موجب زوال شناختی ناشی از پارکینسون (PD dementia) شوند. علاوه بر این، دیسبیوز رودهای (gut dysbiosis) یا روده نشتپذیر (leaky gut) ممکن است موجب فعالسازی سیستم ایمنی (immune system activation) ــ از جمله میکروگلیا به عنوان ماکروفاژهای ساکن مغز (microglia as brain-resident macrophages) ــ شود و در پاتوژنز آلزایمر و پارکینسون نقش داشته باشد. پیکانها در شکل نشاندهنده تحریک (stimulation) هستند و علامتهای T شکل نشانگر مهار (inhibition) میباشند.
میان تغییرات اپیژنتیکی گوناگون (epigenetic alterations) و اختلالات سیناپسی (synaptic dysfunctions) و اختلالات شناختی (cognitive dysfunctions) مرتبط با بیماری آلزایمر (AD) رابطهای نزدیک وجود دارد. به عنوان نمونه، مشخص شده است که NEAT1 ــ یک RNA بلند غیرکدکننده (long noncoding RNA; lncRNA) ــ در تنظیم بیان ژنهای مرتبط با اندوسیتوز آمیلوئید بتا (Aβ endocytosis-related genes) از طریق سازوکارهای اپیژنتیکی (epigenetic mechanisms) نقش دارد، و کاهش بیان آن (downregulation) سبب اختلال در جذب آمیلوئید بتا (Aβ uptake) از راه تنظیم فعالیتهای رونویسی این ژنها (transcriptional activities) میشود [27]. از آنجا که خوداستیلاسیون (autoacetylation) آنزیم P300 و فعالیت استیلترانسفراز (acyltransferase) توسط NEAT1 میانجیگری میشوند، این lncRNA قادر است کروتونایلاسیون (crotonylation) و استیلاسیون هیستون 3 در لیزین 27 (histone 3 lysine 27 acetylation; H3K27ac) را در نواحی مجاور محل آغاز رونویسی (transcription start sites) تغییر دهد. این تغییرات بر اتصال فاکتور رونویسی STAT3 (transcriptional factor STAT3) به این ژنها برای فعالسازی رونویسی (triggering transcription) تأثیر میگذارند [27].
نمونههای دیگر عبارتاند از:
افزایش متیلاسیون DNA (DNA methylation) در ژن TREM2 در ژیروس تمپورال فوقانی (superior temporal gyrus) بیماران مبتلا به آلزایمر [28]،
کاهش متیلاسیون DNA (DNA hypomethylation) در ژن Complement C3 در مغزهای پس از مرگ (postmortem brains) بیماران آلزایمری [29]،
متیلاسیون غیرطبیعی DNA (aberrant DNA methylation) در بیش از ۴۰۰ ژن ــ بهویژه هیپومتیلاسیون پروموتر ژن TGFB3 (TGFB3 promoter hypomethylation) ــ در تحلیل متیلاسیون ژنومی کامل قشر تمپورال (whole-genome DNA methylation analysis of temporal cortex) بیماران آلزایمری [30]،
تغییر متیلاسیون DNA در ۵۸۹۷ ناحیه پروموتر ژنی در ژیروس دندانهدار (dentate gyrus) بیماران آلزایمری، بهویژه هیپومتیلاسیون پروموتر ژن PEN2 (PEN2 gene promoter hypomethylation) [31]،
تغییر متیلاسیون DNA در چندین سایت CpG (حدود ۱۲ ناحیه) که شش مورد از آنها رابطه معکوس با بیان ژنی متناظر خود دارند (برای مثال: TENT5A، PRKCZ و DIRAS1) در قشر انتورینال (entorhinal cortex) بیماران پارکینسونی [32]،
و در نهایت، هیپومتیلاسیون DNA ژن APP (DNA hypomethylation of APP) در بیماران آلزایمری [33] که با رسوب آمیلوئید بتا (Aβ deposition) مرتبط است.
افزون بر این، پژوهشگران دیگر یافتههای بیشتری درباره تغییرات هیستونی (histone modifications)، میکرو RNA (miRNA) و تغییرات اپیژنتیکی اختصاصی سلول (cell-specific epigenetic changes) در زمینه پیری و آلزایمر (aging and AD) گزارش کردهاند [34–36].
شایان توجه است که بخش بزرگی از تغییرات اپیژنتیکی (epigenetic alterations) در بیماری آلزایمر (AD) در سلولهای غیرنورونی (non-neuronal cells) رخ میدهد [37]؛ سلولهایی که در پاکسازی آمیلوئید بتا (Aβ clearance) نقش دارند. برای مثال، مطالعهای اخیر نشان داده است که ایریسین (irisin) ــ هورمونی که در اثر فعالیت بدنی (physical exercise) تولید میشود ــ با فعالسازی گیرندههای آستروسیتی خود (astrocytic receptors) یعنی اینتگرین αV/β5 (integrin αV/β5)، موجب کاهش مسیر پیامرسانی ERK–STAT3 (downregulation of ERK–STAT3 signaling) و افزایش ترشح آنزیم نپریلیزین (neprilysin; NEP) ــ که تجزیهکننده آمیلوئید بتا (Aβ-degrading enzyme) است ــ میگردد، و در نهایت باعث پاکسازی آمیلوئید بتا (Aβ clearance) میشود [38]. آنزیم NEP شناخته شده است که توسط تعدیلات اپیژنتیکی (epigenetic modifications) تنظیم میشود [39]. با این حال، تأثیر فعالیت بدنی بر تعدیل اپیژنتیکی NEP (NEP epigenetic modulation) هنوز بهطور کامل آشکار نشده است. مطالعات کنونی نشان میدهند که ناهنجاریهای اپیژنتیکی (epigenetic aberrations) میتوانند در پاسخ به رسوب آمیلوئید بتا (Aβ deposition) و تجمع تاو (tau accumulation) نیز رخ دهند. برای نمونه، افزایش بیان HDAC4 (increase in HDAC4 expression) در پاسخ به رسوب آمیلوئید بتا در مدل موشی آلزایمر (mouse model of AD) مشاهده شده است [40]، و همچنین کاهش سطح استیلاسیون لیزین 9 هیستون H3 (reduced acetylation level of lysine 9 of histone H3; H3K9ac) در مدل مخمری با بیان بیش از حد آمیلوئید بتا (yeast Aβ overexpression model) گزارش شده است [41]. سایر شواهدی که ارتباط میان تغییرات اپیژنتیکی و رسوب آمیلوئید بتا (Aβ deposition) ــ به عنوان یکی از ویژگیهای شاخص آلزایمر (hallmarks of AD) ــ را نشان میدهند، در جدول 1 (Table 1) خلاصه شدهاند.
جدول 1. تغییرات اپیژنتیکی (Epigenetic alterations) در بیماری آلزایمر (Alzheimer’s disease; AD) که بر رسوب آمیلوئید بتا (amyloid-β deposition) تأثیر میگذارند یا از آن تأثیر میپذیرند.
| تغییرات اپیژنتیک | نوع پروتئین تجمعی | نتایج کلیدی | مرجع |
|---|---|---|---|
| متیلاسیون DNA (DNA methylation) | رسوب آمیلوئید بتا (Aβ deposition) | افزایش متیلاسیون DNA در ژن NXN در شرایط in vitro | [42] |
| متیلاسیون DNA | رسوب آمیلوئید بتا | بیش از 5000 تغییر متیلاسیون DNA در نورونهای dentate gyrus در بیماران آلزایمر (AD) | [31] |
| متیلاسیون DNA | رسوب آمیلوئید بتا | – شناسایی 409 ژن با متیلاسیون غیرطبیعی که در تنظیم تشکیل آمیلوئید بتا نقش دارند– کاهش متیلاسیون پروموتر TGFBR3 در بیماران AD | [30] |
| متیلاسیون DNA | رسوب آمیلوئید بتا | هیپومتیلاسیون ژن APP در بیماران AD | [33] |
| متیلاسیون DNA | تجمع تائو هایپرفسفریله (Accumulation of hyperphosphorylated tau) | ارتباط بین متیلاسیون DNA در چندین CpG از ژن HOXA5 و سطوح pTau181 در مایع مغزی-نخاعی (CSF) | [43] |
| متیلاسیون DNA | تائوی هایپرفسفریله و تائوی تام در CSF | متیلاسیون پروموتر TOMM40 و جزیره CpG ژن APOE در مغز با P-tau و total tau در CSF بیماران AD همبستگی دارد | [44] |
| متیلاسیون DNA/هیستون | رسوب آمیلوئید بتا (Aβ1–42) | ارتباط مستقیم بین افزایش سطوح Aβ42 و افزایش متیلاسیون DNA و هیستون ژن Igf2 در موشهای 5xFAD و بیماران AD | [45] |
| استیلاسیون هیستون (Histone acetylation) | تجمع تائوی هایپرفسفریله | – تغییر در ساختار کروماتین در نورونهای مشتق از سلولهای بنیادی پرتوان القایی (iPSC-derived neurons)– تغییر در استیلاسیون هیستون H3K9ac | [46] |
| استیلاسیون هیستون | رسوب آمیلوئید بتا | افزایش بیان Hdac4 در نتیجه رسوب آمیلوئید بتا در مدل موشی آلزایمر | [40] |
| استیلاسیون هیستون | رسوب آمیلوئید بتا | کاهش H3K9ac در نتیجه بیان بیش از حد Aβ1–40 در مدل مخمری با بیان بیشازحد Aβ | [41] |
| استیلاسیون هیستون | رسوب آمیلوئید بتا (Aβ1–42) | کاهش سطوح استیلاسیون H3 | [47] |
| استیلاسیون یا متیلاسیون هیستون | رسوب آمیلوئید بتا (Aβ42) | افزایش قابلتوجه در استیلاسیون کلی H3 و تریمتیلاسیون H3K27 و H3K9 در مدل مگس میوه (Drosophila) آلزایمر | [48] |
| متیلاسیون هیستون | رسوب آمیلوئید بتا | افزایش بیان G9a (آنزیم لیزین متیلترانسفراز) موجب افزایش دیمتیلاسیون H3K9 در C. elegans میشود | [49] |
| میکروRNAها (MicroRNAs) | رسوب آمیلوئید بتا | افزایش بیان miR-26b در سلولهای N2a/APP موجب کاهش بیان پروتئین Igf-1 و افزایش تولید Aβ در مدل موشی دوترانسژنیک آلزایمر میشود | [50] |
| میکروRNAها | رسوب آمیلوئید بتا | افزایش بیان microRNA-206 موجب افزایش التهاب القاشده با LPS و افزایش آزادسازی Aβ در میکروگلیا از طریق هدفگیری ناحیه 3′-UTR ژن Igf-1 میشود | [51] |
| میکروRNAها | رسوب آمیلوئید بتا | ارتباط بین کاهش miR-137، miR-181c، miR-9 و miR-29a/b-1 با افزایش آنزیم serine palmitoyl-transferase و در نتیجه افزایش سطح Aβ در مدل موشی | [52] |
| میکروRNAها | رسوب آمیلوئید بتا | همبستگی مستقیم بین میکروRNAهای عروقی و پاکسازی آمیلوئید بتا در موشهای جوان 3xTg-AD | [53] |
AD: Alzheimer’s disease; APP: Aβ precursor protein; LPS: Lipopolysaccharide.
پارکینسون (PD)، تجمع آلفا-سینوکلئین و ارتباط آن با تغییرات اپیژنتیکی (epigenetic changes)
بیماری پارکینسون (Parkinson’s disease; PD) به عنوان دومین بیماری نورودژنراتیو مرتبط با سن (age-related neurodegenerative disease) شایع شناخته میشود و معمولاً ۱–۴٪ از افراد بالای ۶۰ سال را تحت تأثیر قرار میدهد؛ بیش از ۵ میلیون نفر در سراسر جهان به این بیماری مبتلا هستند [54,55]. پارکینسون با اختلالات سیناپسی و حرکتی (synaptic and motor dysfunction) و اختلالات شناختی (cognitive impairment) مشخص میشود که طیف آن از اختلال شناختی خفیف (mild cognitive impairment) تا زوال شناختی ناشی از پارکینسون (PD dementia) متفاوت است [56]. ویژگیهای شاخص پارکینسون (hallmarks of PD) شامل تجمع گسترده پروتئین آلفا-سینوکلئین (α-synuclein protein aggregation) در قالب اجسام لوی (Lewy bodies) و از بین رفتن نورونهای دوپامینرژیک (dopaminergic neuronal loss) در ناحیه سیاه متراکم (substantia nigra pars compacta) میباشد (شکل 1) [57,58]. سیستم ایمنی ذاتی (innate immune system) توسط تجمعات پروتئینی نادرست تاخورده (misfolded protein aggregates) در هر دو بیماری آلزایمر و پارکینسون فعال میشود که این امر موجب آغاز التهاب (initiation of inflammation) و تشدید آسیب بافتی و اختلال عملکرد سلولی (exacerbating tissue damage and cellular dysfunction) میگردد [59,60]. فعالسازی بیشازحد سیستم ایمنی ذاتی (overstimulation of the innate immune system) از طریق سیگنالدهی گیرندههای نوع Toll (Toll-like receptor signaling) میتواند توسط دیسبیوز رودهای (gut dysbiosis) رخ دهد و در نتیجه منجر به التهاب موضعی و سیستمیک (local as well as systemic inflammation) و توسعه پاتوژنی آلفا-سینوکلئین (α-synuclein pathology) شود [61,62].
تجمع خارجسلولی آلفا-سینوکلئین (extracellular α-synuclein aggregation) در بیماری پارکینسون (PD) با تغییرات اپیژنتیکی (epigenetic alterations) مرتبط است. گزارش شده است که آلفا-سینوکلئین (α-synuclein) میتواند به طور مستقیم به پروتئینهای هیستون (histone proteins) متصل شود و موجب کاهش سطح هیستون H3 استیلهشده (acetylated histone H3 level) در مدلهای سلولی PD گردد و فرایند استیلاسیون در آزمایشهای هیستون استیلترانسفراز (histone acetyltransferase assays) را مختل کند [63]. بنابراین، مهارکنندههای هیستون دهیدروژناز (histone deacetylase inhibitors) ممکن است نامزدهای بالقوه برای کاهش سمیت آلفا-سینوکلئین (α-synuclein toxicity) در بیماران پارکینسونی باشند [64,65]. رونویسی ژن آلفا-سینوکلئین (α-synuclein) که توسط ژن SNCA کد میشود میتواند توسط لیگاندهای β2-آدرنرژکتور (β2-adrenoreceptor ligands) از طریق H3K27ac پروموتر و افزایشدهندههای آن (promoter and enhancers) تنظیم شود [66]. علاوه بر فعالسازی میکروگلیا (microglial activation) و نورودژنراسیون دوپامینرژیک (dopaminergic neurodegeneration)، توسعه PD ممکن است با هایپر استیلاسیون هیستون (histone hyperacetylation) نیز مرتبط باشد. میکروگلیا فعال شده در مدلهای PD (activated microglia) نشاندهنده هایپر استیلاسیون هیستون (histone hyperacetylation) است و مهارکنندههای هیستون دهیدروژناز (HDAC inhibitors) میتوانند نورودژنراسیون و فعالسازی میکروگلیا را کاهش دهند (mitigate neurodegeneration and microglial activation) [67]. همچنین، نورودژنراسیون در طول پیشرفت PD به اختلال در انتقال هستهای–سیتوپلاسمی HDAC1 (impaired nucleocytoplasmic translocation of HDAC1) و در نتیجه مختل شدن استیلاسیون هیستون (disturbed histone acetylation) نسبت داده شده است [68]. در همین راستا، مطالعهای دیگر نشان داد که استیلاسیون افزایشیافته در بسیاری از سایتهای هیستونی (increased acetylation of many histone sites) و هایپر استیلاسیون گسترده H3K27 در سراسر ژنوم (genome-wide hyperacetylation of H3K27)، از جمله در ژنهای مرتبط با PD (e.g., SNCA, PARK7, PRKN and MAPT)، در بافتهای مغزی پس از مرگ بیماران PD (postmortem brain tissues of PD patients) مشاهده شده است [69].
علاوه بر تغییرات هیستونی (histone modifications)، هیپومتیلاسیون DNA (DNA hypomethylation) در سایتهای CpG داخل اینترون ۱ ژن SNCA (SNCA intron 1 CpG sites) در سلولهای خونی (blood cells) [70] و مغزهای پس از مرگ بیماران پارکینسونی (postmortem brains of PD patients) مشاهده شده است، بهویژه در افرادی که جهش GBA1 دارند [71]. همچنین هیپومتیلاسیون DNA پروموتر SNCA (SNCA promoter) در سلولهای نمونه بزاق (saliva samples) گروهی از افراد آفریقایی، به ویژه در بیماران با پارکینسون خانوادگی (familial PD) گزارش شده است [72]. تحلیل متیلاسیون ژنومی کامل (whole-genome DNA methylation analysis) همچنین تغییر متیلاسیون در ژنهای NFYA, DDR1, RNF5, AGPAT1 (یک استیلترانسفراز; acetyltransferase) و vault RNA VTRNA2-1 [73] و همچنین حدود دو دوجین ژن دیگر (مانند هیپومتیلاسیون CACNA1B, CREB5, GNB4 و PPP2R5A) در سلولهای خونی بیماران PD [74] و صدها ژن در سلولهای نوروبلاستوم تحت تأثیر 6-OHDA (مانند هایپرمتلاسیون ABHD5, FADS3 و UGT2A2 و هیپومتیلاسیون RAD51B, DOCK5 و GRK5) [75] شناسایی شد. مطالعهای دیگر گزارش کرده است که افزایش متیلاسیون DNA ژنهای EFEMP1 و CD56 در سلولهای خونی با کاهش و افزایش ریسک PD به ترتیب مرتبط است [76]. علاوه بر این، تحلیل متیلاسیون ژنومی کامل مغزهای پس از مرگ (whole-genome DNA methylation) از نمونههای قشر پیشپیشانی (prefrontal cortex) بیماران PD با زوال شناختی (dementia)، الگویی متمایز و بهطور برجسته هیپومتیلهشده (82٪ از CpGها) را در مقایسه با دیگر انواع زوال شناختی و افراد کنترل نشان داد [77]. همچنین شایان ذکر است که تحلیل اپیژنتیکی دیگر نشان داد که از ۵۲۱ ژن مسیر اتوفاژی–لیزوزوم (autophagy–lysosome pathway)، ۳۲۶ ژن دارای متیلاسیون DNA غیرطبیعی (اغلب هایپرمتیلهشده) در آپاندیس بیماران PD بودند. مطالعات تکمیلی در موشها نشان داد که التهاب مزمن روده (chronic gut inflammation) میتواند تغییرات اپیژنتیکی این ژنها را القا کند و پاتولوژی آلفا-سینوکلئین (α-synuclein pathology) میتواند آن را تشدید نماید [78]. علاوه بر این، گزارشهایی نیز درباره اختلال در miRNAهای اگزووزومی (exosomal miR-501-3p, miR-126-5p و miR-99a-5p) در مایع مغزی-نخاعی (CSF) [79]، ۳۴ lncRNA در مغزهای پس از مرگ [80] و صدها RNA دایرهای (circular RNAs) در خون بیماران PD [81] منتشر شده است. در پارکینسون با شروع زودرس (early onset PD)، تغییر متیلاسیون بیش از ۲۰۰۰ ژن (اغلب هایپرمتیلهشده و مرتبط با عملکرد نورونی و پاسخ ایمنی) در DNA سلول-آزاد CSF (cell-free DNA of CSF) مشاهده شد [82]. جدول 2 (Table 2) خلاصهای از این دادهها و شواهد دیگر را ارائه میدهد که ارتباط میان ناهنجاریهای اپیژنتیکی متنوع و افزایش بیان یا تجمع آلفا-سینوکلئین (α-synuclein) در PD را نشان میدهند.
جدول 2. تغییرات اپیژنتیکی (Epigenetic alterations) در بیماری پارکینسون (Parkinson’s disease; PD) که بر بیان یا تجمع آلفا-سینوکلئین (α-synuclein expression or accumulation) تأثیر میگذارند یا از آن تأثیر میپذیرند.
| تغییرات اپیژنتیک | نتایج کلیدی | مرجع |
|---|---|---|
| Histone acetylation and methylation | کنترل بیان SNCA در PD از طریق تغییرات histone در SNCA؛ افزایش H3K4me3 پروموتر SNCA در substantia nigra در PD | [83] |
| Histone methylation | کاهش H3K27me3 و Ezh2 با ubiquitination و تجزیه پروتئازومی α-synuclein در مدل موشی PD مرتبط است | [84] |
| DNA methylation | DNA hypomethylation در intron-1 SNCA باعث افزایش بیان α-synuclein در بیماران sporadic PD میشود | [85] |
| DNA methylation | DNA hypomethylation در سایتهای CpG intron 1 SNCA در AD مشاهده شد | [69] |
| Histone ubiquitylation | کاهش monoubiquitylation histone 2A در lysine 119 توسط hyperphosphorylation زیرواحد BMI-1 و تجمع سلولی phosphorylated α-synuclein در serine 129 در سلولهای Rotenone-treated SH-SY5Y مرتبط است | [86] |
| Histone acetylation | ارتباط مستقیم بین histone H3 acetylation پروموتر Snca و بیان α-synuclein در موشهای تزریقشده با MPTP | [87] |
| MicroRNA expression | اختلال در بیان ۳۲ miRNA و ۱۱۲ piRNA در Caenorhabditis elegans ترنسژنیک با α-synuclein جهشیافته انسانی نسبت به α-synuclein نوع وحشی | [9] |
| MicroRNA expression | تنظیم محور Bdnf/α-synuclein در مراحل اولیه PD توسط miRNA-7 در موشهای PD القا شده با atrazine | [88] |
| MicroRNA expression | افزایش miR-7 و miR-30 در سلولهای blood mononuclear بیماران PD | [88] |
| MicroRNA expression | افزایش miR-101a-3p (به عنوان synaptic miRNA) در موشهای ترنسژنیک α-synuclein و قشر مغزی بیماران با dementia و تجمع Lewy body | [89] |
| Long noncoding RNA | α-synuclein باعث افزایش بیان IL6ST-AS، یک lncRNA که فعالسازی microglia و نکروز نورونی در سلولهای SH-SY5Y را القا میکند | [90] |
| Histone acetyl-transferase (HAT) expression | کاهش سطح histone acetyltransferase در مغز موشهای PD که با مهار HDAC درمان شده و باعث کاهش α-synuclein و inflammatory cytokines میشود | [91] |
| DNA hydroxymethylation and histone H3K27ac | تاثیر بر CpG hydroxymethylation هیپوکامپ و histone H3K27ac در موشهای ترنسژنیک | [91] |
| DNA methylation | Hypomethylation SNCA در leukocytes بیماران PD | [92] |
| DNA methylation | افزایش سطح 5-hmC در cerebellar white matter و افزایش سطح 5-mC SNCA در بخشهای قشری مغز بیماران PD | [93] |
| MicroRNA expression | افزایش miR-19a-3p در exosomes سلولهای ترنسژنیک SH-SY5Y با تجمع α-synuclein و اختلال autophagy در microglia گیرنده | [94] |
| MicroRNA expression | افزایش بیان miRNA-384-5p که پیشرفت PD را با هدفگیری SIRT1 در سلولهای SH-SY5Y و موشها ارتقا میدهد | [95] |
5-mC: 5-methylcytosine; 5-hmC: 5-hydroxymethylcytosine; AD: Alzheimer’s disease; HDAC: Histone deacetylase; MPTP: 1-methyl-4-phenyl-1,2,3,6-tetrathydropyridine; PD: Parkinson’s disease; SNCA: α-synuclein.
روده نشتپذیر (Leaky gut) و نقش آن در آلزایمر و پارکینسون از طریق التهاب و مکانیسمهای اپیژنتیکی (inflammation & epigenetic mechanisms)
روده نشتپذیر (Leaky gut) به وضعیتی اطلاق میشود که در آن لایه پوششی روده (intestinal lining) نفوذپذیری بیشتری پیدا میکند (more permeable) و اجازه میدهد مواد مضر (harmful substances) از جمله توکسینهای باکتریایی (bacterial toxins)، متابولیتهای سمی گوارشی (toxic digestive byproducts)، مولکولهای کوچک (small molecules) و باکتریهای بیماریزا (pathogenic bacteria) وارد جریان خون (bloodstream) شوند [96]. نفوذپذیری پارانشیمی مخاط روده (intestinal mucosal paracellular permeability) میتواند تحت تأثیر عوامل متعددی از جمله استرسهای فیزیولوژیک (physiological stressors)، اضطراب (anxiety)، ترکیبات غذایی مانند امولسیفایرها (dietary components; emulsifiers) و تمرینات فیزیکی شدید (intensive physical exercise) تغییر کند. این عوامل باعث افزایش ورود باکتریها و توکسینهای باکتریایی به گردش خون سیستمیک (systemic circulation) میشوند، که التهاب سیستمیک (systemic inflammation) را تحریک میکند و بهتبع آن ممکن است نفوذپذیری سد خونی–مغزی (BBB permeability) تغییر کند و عملکرد مغز (brain functions) را تحت تأثیر قرار دهد [97]. شایان ذکر است که تغییرات مرتبط با سن در نفوذپذیری روده و عملکردهای ایمنی (age-related alterations in intestinal permeability and immune functions) نیز ممکن است فرصت ورود باکتریهای رودهای به جریان خون (gut bacteria into blood circulation) را فراهم کنند [98]. جالبتر اینکه، تحلیل اخیر آمپلیکون 16S rRNA (16S rRNA amplicon analysis) تفاوتهای چشمگیری در ترکیب میکروبیوتای خون (blood microbiota composition) بین بیماران پارکینسونی (PD patients) و افراد کنترل همسن (age-matched control subjects) نشان داد [99].
التهاب خفیف (Low-grade inflammation) در افراد مسن (older adults) با افزایش نفوذپذیری اپیتلیوم رودهای (enhanced intestinal epithelial permeability) و میانجیگری میکروبیوتای غیرطبیعی روده (abnormal gut microbiota) مرتبط است. مطالعهای توسط Forsyth و همکارانش نشان داد که افزایش نفوذپذیری روده در افرادی که به تازگی با پارکینسون تشخیص داده شدهاند (newly diagnosed PD subjects) با افزایش رنگآمیزی مخاط روده برای باکتریهای Escherichia coli، نیتروتیروزین (nitrotyrosine)، آلفا-سینوکلئین (α-synuclein) و سطح سرمی پروتئین اتصالدهنده لیپوپلیساکارید (serum lipopolysaccharide binding protein) همراه است [100]. همچنین گزارش شده است که تغییرات ایمنی سیستمیک (systemic immunological alterations) ناشی از عفونت با Helicobacter pylori در معده نقش کلیدی در نورواِنتفلاماسیون (neuroinflammation) و بنابراین آغاز آلزایمر و پارکینسون (onset of AD and PD) ایفا میکنند [101]. علاوه بر این، در مدل موشی APP/PS1 آلزایمر (APP/PS1 mouse model of AD) با روده نشتپذیر و بدون آن (with and without leaky gut)، Yadav و همکارانش مشاهده کردند که افزایش نفوذپذیری روده (leaky gut) با زوال شناختی (cognitive decline) و سطوح بالاتر نشانگرهای مغزی آلزایمر (brain markers of AD) مرتبط است، اما این اثرات با تغذیه با پروبیوتیکهای تازه جداسازیشده از روده نوزاد (newly isolated probiotics from infant’s gut) کاهش مییابد [102]. نتایج آنها نشان داد که اثرات مفید تغذیه با پروبیوتیکها در کاهش تجمع آمیلوئید بتا (amyloid-β accumulation)، زوال شناختی و التهاب (inflammation) با کاهش نشت روده (reduced gut leakiness) و افزایش تولید متابولیتهای مفید (enhanced production of beneficial metabolites) مرتبط بوده است.
یکی از فرضیات پاتوژنز آلزایمر به نام «فرضیه پاتوژن (pathogen hypothesis)» یا «فرضیه عفونی (infection hypothesis)» شناخته میشود، که در آن عفونتهای مزمن باکتریایی، ویروسی یا قارچی (chronic bacterial, virus or fungal infections) به عنوان عامل شروع آلزایمر اسپادیک (sporadic AD) در طول پیری (aging) در نظر گرفته میشوند [103]. گفته شده است که عفونتهای میکروبی مزمن (chronic microbial infections) موجب فعالسازی سیستم ایمنی (immune system activation) و التهاب مزمن (chronic inflammation) میشوند، که این امر فرصت عبور پاتوژنهای میکروبی و/یا محصولات آنها (microbial pathogens and/or their products) از سد خونی–مغزی (BBB; blood–brain barrier) را فراهم میکند [96]. در این راستا، در موشهای مدل آلزایمر (AD model rodents)، تعامل نزدیک (close interplay) بین تغییرات ترکیب میکروبیوتای روده (altered gut microbiota composition)، از دست رفتن پروتئینهای اتصال محکم و اتصال چسبنده (tight junction and adherence junction proteins)، افزایش آستروگلیوز (elevated astrogliosis) و فعالسازی میکروگلیا (microglial activation) و همچنین افزایش بیان اینفلاماسوم NLRP3 همراه با ترشح IL-1β در بافت مغزی (stronger expression of NLRP3 inflammasome along with IL-1β secretion in brain tissue) مشاهده شده است [104]. علاوه بر این، عفونتهای میکروبی (microbial infections) با تولید آمیلوئید بتا (Aβ production) و تشکیل پلاکهای سالمندی (senile plaques) نیز مرتبط هستند.
تأثیرات منفی باکتریها (bacteria) بر عملکرد مغز (brain functions) و آغاز آلزایمر (onset of AD) شامل تغییرات اپیژنتیکی (epigenetic alterations) میشود. برای مثال، مشخص شده است که برخی باکتریها مانند Chlamydia pneumoniae، یک پاتوژن داخلسلولی گرم-منفی (intracellular Gram-negative pathogen)، قادرند با پروتئینهای هیستونی میزبان (host histone proteins) تعامل کنند و سیستم تنظیم اپیژنتیکی میزبان (host’s epigenetic regulatory system) را پس از ورود محصولات باکتریایی به مغز (bacterial product intrusion into the brain) تغییر دهند و از این طریق آغاز آلزایمر (onset of AD) را از طریق ایجاد پاسخ التهابی پایدار (persistent inflammatory response) القا کنند [105]. در مقابل، پروبیوتیکها (probiotics) به عنوان نامزدهای مناسب برای کاهش روده نشتپذیر مرتبط با پیری و پاسخهای التهابی (aging-related leaky gut and inflammatory responses) شناخته میشوند. به عنوان مثال، Yadav و همکارانش اثرات محافظتی کوکتل پروبیوتیکی با منشاء انسانی (human-origin probiotic cocktail) شامل پنج سویه Enterococcus و پنج سویه Lactobacillus (از روده نوزادان سالم جداسازی شده) را در برابر روده نشتپذیر و التهاب مرتبط با پیری بررسی کردند. آنها دریافتند که این ترکیب پروبیوتیکی (probiotic composition) قادر است دیسبیوز میکروبیوتای روده ناشی از رژیم غذایی پرچرب (high-fat diet-induced gut microbiota dysbiosis) را مهار کند و روده نشتپذیر (leaky gut) را با تقویت اتصالات محکم (tight junctions) کاهش دهد، که به نوبه خود التهاب را تخفیف میدهد (attenuate inflammation) [106]. همچنین نشان داده شده است که اسیدهای چرب کوتاهزنجیر (short-chain fatty acids; SCFAs)، متابولیتهای تولیدشده توسط باکتریهای دستگاه گوارش (GI bacteria) از طریق تخمیر فیبر غذایی (fermentation of dietary fiber)، به عنوان تغییردهندههای اپیژنتیکی قوی (powerful epigenetic modifiers) نقش مهمی در حفظ یکپارچگی سد رودهای (gut barrier integrity) و هومئوستازی روده (homeostasis in the intestine) ایفا میکنند. به دنبال آن، این متابولیتها با نرمال کردن سطح استیلاسیون هیستون هیپوکامپ مغز (brain hippocampal histone acetylation levels) از اختلالات شناختی نورونی در موشهای مسن (neurocognitive deficits in aged mice) جلوگیری میکنند [107]. به عنوان مثال، بوتیرات (butyrate) توانایی کاهش نفوذپذیری بیش از حد روده و التهاب مرتبط با روده نشتپذیر (intestinal hyperpermeability and inflammation relevant to leaky gut) را از طریق افزایش بیان پروتئینهای اتصال محکم رودهای (expression of intestinal tight junction proteins) نشان داده است [108].
عملکرد مغز (brain functions) و رفتار (behavior) تحت تأثیر میکروبیوتای روده (gut microbiota) قرار دارند، که از طریق مسیرهای ایمنی، عصبی و اندوکرینی (immunological, neurological and endocrine pathways) یک سیستم چندجهتی و پیچیده (complex multidirectional system) را شکل میدهند که به آن محور میکروبیوم–روده–مغز (microbiome–gut–brain axis) گفته میشود. حمایت از این ایده با تغییرات چشمگیر در میزان و ترکیب باکتریهای روده و متابولیتهای آنها (abundance and composition of gut bacteria and their metabolites) در بیماران پارکینسون (PD patients) گزارش شده است [109]. همچنین سطح بالای تریمتیلآمین N-اکسید (trimethylamine N-oxide; TMAO)، یک مولکول کوچک (small molecule) و متابولیت مشتق از میکروبیوتای روده (gut microbiota-derived metabolite) که از متابولیسم کولین رژیمی (metabolism of dietary choline) تولید میشود—که منبع اصلی گروه متیل (main source of methyl group) است—در افراد مبتلا به آلزایمر (AD) و مرتبط با تائو فسفریلهشده و نسبت تائو فسفریلهشده به Aβ42 (phosphorylated tau and phosphorylated tau/Aβ42) گزارش شده، در مقایسه با افراد بدون اختلال شناختی (cognitively unimpaired individuals) [110]. بنابراین، سایر تغییرات گزارششده در میکروبیوتای روده (other reported alterations in gut microbiota)، مانند کاهش Enterococcaceae و Lactobacillaceae (دو خانواده باکتریایی از شاخه Firmicutes) و افزایش خانواده باکتریایی Enterobacteriaceae، همراه با تغییرات در تولید SCFA در نمونههای مدفوع بیماران PD نسبت به افراد کنترل مطابقت داده شده (matched controls) [109] میتواند تأثیرات مشابهی بر عملکرد مغز و بیماریهای عصبی (similar effects) داشته باشد.
تغییرات میکروبیوم روده (intestinal microbiome alterations) نه تنها به تجمع آلفا-سینوکلئین (α-synuclein accumulation) کمک میکنند، بلکه پاسخ التهابی در بافتهای محیطی (inflammatory response in peripheral tissues) را نیز تحریک میکنند، از جمله فعالسازی سلولهای T (T cell activation) و ترشح بیش از حد سایتوکینها (excessive cytokine secretion). به نظر میرسد که عصب واگ (vagus nerve) مسئول انتشار تجمع آلفا-سینوکلئین به سبک پریون (prion-like propagation) است، که ابتدا از محیط پیرامونی (vagus nerve in the medulla) به سیستم عصبی مرکزی (central nervous system) منتقل میشود [111]. مطالعه Sampson و همکارانش نیز نقش میکروبهای روده (gut microbes) در نوروتوکسیسیتی و التهاب ناشی از α-synuclein (α-synuclein-induced neurotoxicity and inflammation) را تأیید میکند. در این مطالعه، مشاهده شد که اختلالات حرکتی (motor impairment) در موشهای PD-like فاقد میکروب (germ-free PD-like mice) افزایش یافت، زمانی که میکروبیوتای روده از بیماران PD به آنها منتقل شد (received the gut microbiota from PD patients) [112]. همچنین نشان داده شده است که پاتوژنز PD (PD pathogenesis) با تغییرات میکروبیوتا در خون و مغز (altered microbiota in blood and brain) مرتبط است، زیرا کاهش فراوانی جنس Blautia (Blautia genus) که عملکرد میتوکندری و متابولیسم انرژی (mitochondrial functions and energy metabolism) را تحت تأثیر قرار میدهد، در نمونههای خون، مدفوع و مغز بیماران PD نسبت به افراد کنترل مشاهده شد [113]. شایان ذکر است که بر اساس گفته نویسنده، وجود باکتری در خون و مغز ناشی از آلودگی (contamination) نبوده است. در این زمینه، یک مطالعه مقطعی اخیر (recent cross-sectional study) تفاوتهای قابل توجهی در پروفایلهای تاکسونومیک میکروبی روده (gut microbial taxonomic profiles) بین افراد با AD پیشبالینی (preclinical AD) و کسانی که شواهدی از AD پیشبالینی ندارند نشان داد، که حاکی است ترکیب میکروبیوم روده میتواند به عنوان شاخصی از AD پیشبالینی (indicator of preclinical AD) عمل کند [114]. مطالعه دیگری ارتباط بین تغییر ترکیب میکروبی، پروفایلهای اپیژنتیکی (epigenetic profiles) و عملکرد رفتاری و شناختی (behavioral and cognitive performance) را در مدل موشی AD نشان داد. نویسندگان ارتباط مثبت بین متیلاسیون DNA ژن Apoe در هیپوکامپ (DNA methylation of the Apoe gene in the hippocampus) و سویههای خاص آمپلیکون در خانواده Lachnospiraceae (specific amplicon sequence variants within the Lachnospiraceae family) را شناسایی کردند [115].
اسیدهای چرب کوتاهزنجیر (short-chain fatty acids; SCFAs) نقش حیاتی در ارتباط روده و مغز (gut and brain crosstalk) دارند. آنها در چندین فرآیند فیزیولوژیک (physiological processes) مشارکت میکنند، از جمله تنظیم ایمنی ذاتی مخاط روده (modulation of gut mucosal innate immunity)، حفظ یکپارچگی سد رودهای (preservation of intestinal barrier integrity) و جلوگیری از اثرات مخرب بر سد خونی–مغزی (BBB; detrimental effects in the BBB) با افزایش بیان پروتئینهای اتصال محکم (tight junction proteins) [116]. مطالعات دیگر نشان دادهاند که تغییرات میکروبیوتای روده در پارکینسون (altered gut microbiota in PD) باعث تغییر غلظت SCFAs در مدفوع و پلاسما (fecal and plasma concentrations of SCFAs) میشود. به عنوان مثال، Lin و همکارانش گزارش کردند که سطح کاهشیافته SCFAs مدفوعی خاص (e.g., acetic, propionic and butyric acids) و افزایش سطح پلاسما از پروپیونیک و بوتیریک اسیدها و همچنین والریک اسید (valeric acid; SCFA) در بیماران PD نسبت به افراد کنترل مشاهده شده است [117]. به نوبه خود، SCFAs بر نورواِنتفلاماسیون ناشی از α-synuclein و مرگ سلولهای عصبی (α-synuclein-induced neuroinflammation and neuronal cell death) تأثیر میگذارند، که این اثر از طریق تنظیم فعالیتهای گیرندههای جفتشده با پروتئین G (G protein-coupled receptor activities) اعمال میشود [118].
یک مطالعه پیشین نشان داد که تجمع آمیلوئید (amyloid deposition) و ناهنجاریهای فراساختاری (ultrastructural abnormalities) در روده مدل موشی آلزایمر (AD mouse model) با تغییر ترکیب میکروبیوتا (altered microbiota composition) و کاهش سطح SCFAs (reduced level of SCFAs) همراه است [119]. یک مطالعه انسانی نیز همبستگی معکوس بین آمیلوئیدوز مغز (brain amyloidosis) و سطح بوتیرات خون (blood butyrate level) و همچنین IL-10، یک سایتوکین ضدالتهابی (anti-inflammatory cytokine) را نشان داد [120]. پاتولوژی AD (AD pathology) نه تنها عملکرد مغز و شناخت (brain function and cognition) را تحت تأثیر قرار میدهد، بلکه سطح SCFAها را از طریق تغییرات میکروبیوتای روده ناشی از تجمع آمیلوئید رودهای (gut microbiota alterations induced by intestinal amyloid deposition) کاهش میدهد [119]. در حالی که برخی SCFAها مانند بوتیریک اسید و والریک اسید (butyric acid and valeric acid) در شرایط آزمایشگاهی (in vitro) تجمع آمیلوئید بتا (Aβ aggregation) را مهار میکنند، اختلال متابولیسم SCFA و کاهش سطح بوتیریک اسید (low butyric acid level) باعث افزایش بیان Hdac4 در هیپوکامپ موشها (high expression of Hdac4 in rat hippocampi) و تأثیر بر H4K8ac, H4K12ac و H4K16ac میشود، که به نوبه خود آپوپتوز نورونی (neuronal apoptosis) را افزایش میدهد [121]. بوتیرات نه تنها به عنوان مهارکننده قوی داخلی HDAC (strong endogenous HDAC inhibitor) عمل میکند، بلکه متیلاسیون DNA (DNA methylation) را نیز تحت تأثیر قرار میدهد [122]. در همین راستا، پروفایلسازی متیلاسیون DNA در نمونههای خون کامل بیماران PD و افراد کنترل (DNA methylation profiling of whole-blood samples) نشان داد که کاهش سطح بوتیرات تولیدشده توسط باکتریها در بیماران PD (reduced levels of bacterially produced butyrate in PD patients) منجر به تغییرات متیلاسیون DNA در لوکوسیتها (DNA methylation alterations in leucocytes) شده و شدت علائم افسردگی در PD (increasing the severity of depressive symptoms in PD) را افزایش میدهد [123].
متابولیتهای مشتق از میکروبیوم روده با اثرات اپیژنتیکی در درمان آلزایمر و پارکینسون (Gut microbiome-derived metabolites with epigenetic effects for treatment of AD & PD)
SCFAs (اسیدهای چرب کوتاهزنجیر) توانایی بالقوهای برای درمان بیماران PD (treatment of PD patients) دارند، زیرا قادرند میکروبیوتای روده را بازسازی کنند (remodeling the gut microbiota)، نفوذپذیری روده را کاهش دهند (reducing intestinal permeability)، ترشح رادیکالهای آزاد اکسیژن را مهار کنند (inhibiting reactive oxygen species release)، فعالسازی میکروگلیا را کاهش دهند (decreasing microglial activation) و التهاب رودهای و در نتیجه نورواِنتفلاماسیون را سرکوب کنند (suppressing intestinal inflammation and hence neuroinflammation). در واقع، مطالعات پیشین کاهش قابل توجه فراوانی باکتریهای ضدالتهابی تولیدکننده بوتیرات (butyrate-producing anti-inflammatory bacteria) شامل Blautia، Coprococcus و Roseburia را در بیماران PD نشان دادهاند [109]. مطالعات اخیر نشان دادهاند که SCFAها (به ویژه بوتیرات، اسید والپروئیک و پروپیونات; butyrate, valproic acid and propionate) که از فیبرهای رژیمی توسط تخمیر باکتریایی (bacterial fermentation of dietary fibers) تولید میشوند، عاملان درمانی مؤثری (good therapeutic agents) برای بازسازی ترکیب میکروبیوتا (reshaping microbiota composition)، تقویت شاتل گلوتامات–گلوتامین آستروسیت–نورون (astrocyte–neuron glutamate–glutamine shuttle)، سرکوب نورواِنتفلاماسیون (suppressing neuroinflammation) و در نتیجه بهبود AD و PD (improving AD and PD) هستند، که این اثرات به دلیل توانایی آنها در مهار HDACها (inhibition of HDACs) میباشد [124,125]. بوتیرات (butyrate)، یک SCFA شناختهشده و تغییردهنده اپیژنتیکی (epigenetic modifier)، توسط بیهوازیهای کولون (anaerobes in the colon) از طریق تخمیر فیبرهای غذایی غیرقابل هضم (fermentation of indigestible dietary fibers) تولید میشود و دارای اثرات آنتیاکسیدانی و ضدالتهابی (antioxidant and anti-inflammatory effects) است که در تنظیم میکروبیوتای روده (intestinal microbiota regulation) و ترمیم سد رودهای (intestinal barrier repair) نقش دارد [126,127]. مطالعات دیگر که فواید SCFAها یا داروهای با مکانیزم مشابه (drugs with similar mechanisms of action) برای AD از طریق تغییرات اپیژنتیکی را تأیید میکنند در جدول 3 (Table 3) و مطالعات مربوط به فواید SCFAها یا داروهای مشابه در PD از طریق تغییرات اپیژنتیکی در جدول 4 (Table 4) خلاصه شده است.
جدول 3. اثرات مفید اسیدهای چرب کوتاهزنجیر (SCFAs) یا داروهای مؤثر بر استیلدار شدن هیستونها (histone acetylation) در بیماری آلزایمر (Alzheimer’s disease)
| SCFA یا داروها | نوع مطالعه | نتایج کلیدی | مرجع |
|---|---|---|---|
| سودیم بوتیرات (Sodium butyrate) | مدل موشی 5xFAD بیماری آلزایمر (5xFAD mouse model of AD) | کاهش ۴۰٪ سطح Aβ مغز و افزایش ۲۵٪ پاسخ ترس در مدل موشی 5xFAD پس از ۱۲ هفته تغذیه با سودیم بوتیرات | [128] |
| سودیم بوتیرات (Sodium butyrate) | سلولهای SH-SY5Y تحریکشده با TNF-α، سلولهای BV-2 تحریکشده با LPS و سلولهای SH-SY5Y القاشده با Aβ (TNF-α-stimulated SH-SY5Y, LPS-induced BV-2 and Aβ-induced SH-SY5Y cells) | کاهش هیپرفسفریلاسیون tau در سلولهای SH-SY5Y تحریکشده با TNF-α و BV-2 القاشده با LPS و همچنین مهار ROS در سلولهای SH-SY5Y القاشده با Aβ | [129] |
| سودیم بوتیرات (Sodium butyrate) | مدل موشی 5XFAD بیماری آلزایمر (5XFAD mouse model of AD) | مهار فعالسازی بیش از حد میکروگلیا، تجمع Aβ و بهبود پلاستیسیتی سیناپسی با کاهش نورواِینفلامیشن در مراحل اولیه بیماری در موشهای 5XFAD | [130] |
| سودیم بوتیرات (Sodium butyrate) | مدل موشی AD (AD mouse model) | بهبود عملکرد حافظه مرتبط با افزایش میزان استیلاسیون هیستون در هیپوکامپ و افزایش بیان ژنهای مرتبط با یادگیری ارتباطی | [131] |
| سودیم بوتیرات (Sodium butyrate) | مدل موشی AD (AD mouse model) | بهبود عملکرد میتوکندریایی آستروسیتها، افزایش تمایز آستروسیتها به زیرنوع محافظتکننده از نورون A2 و افزایش شاتل لاکتات بین آستروسیتها و نورونها | [132] |
| سودیم والپروات/سودیم بوتیرات (Sodium valproate/sodium butyrate) | مدل موشی AD (AD mouse model) | مهار مشترک Hdac1، ۲، ۳ و ۸ (HDACهای کلاس I) و در نتیجه درمان اختلالات شناختی مرتبط با مراحل اولیه AD | [133] |
| والپرویک اسید (Valproic acid) | آستروسیتهای انسانی و مدل موشی ترانسژنیک AD (Human astrocytes and transgenic AD mouse) | افزایش بیان و ترشح کلسترولین در آستروسیتهای انسانی و پیشگیری از تجمع آمیلوئید-β در موشهای ترانسژنیک AD | [134] |
| سودیم بوتیرات (Sodium butyrate) | نوروتوکسیسیته القاشده با Aβ در سلولهای PC12 (Aβ induced neurotoxicity in PC12 cells) | افزایش بیان آنزیم مبدل آنژیوتانسین و BDNF و فعالسازی GPCRها در برابر نوروتوکسیسیته القاشده با Aβ | [135] |
| سودیم بوتیرات (Sodium butyrate) | آسیب سلولی القاشده با Aβ در سلولهای N2a (Aβ-induced cell damage in N2a cells) | محافظت در برابر آسیب سلولی القاشده با Aβ از طریق فعالسازی GPR109A | [136] |
| سودیم بوتیرات (Sodium butyrate) | مطالعه تجربی در شرایط آزمایشگاهی (Experimental study in vitro) | مهار وابسته به دوز تبدیل مونومرهای Aβ40 به فیبریلهای Aβ | [137] |
| والپرویک اسید (Valproic acid) | مدل موشی AD (AD mouse model) | کاهش tau هیپرفسفریله و تحریک رشد زایدههای نورونی با مهار فعالیت Gsk-3β | [138] |
| والپرویک اسید (Valproic acid) | موشهای Tg6799 MT (Tg6799 MT mice) | بهبود اختلال شناختی از طریق مهار Hdac در موشهای ۵ و ۱۰ ماهه | [139] |
| والپرویک اسید (Valproic acid) | مدل موشی AD اسپورادیک القاشده با استرپتوزوتوسین (Streptozotocin-induced sporadic AD mouse model) | ارتقای سیناپتوژنز و بهبود یادگیری و حافظه از طریق افزایش استیلاسیون هیستون و به دنبال آن افزایش بیان neprilysin (Nep) با باز کردن ساختار کروماتین پروموتر | [140] |
| والپرویک اسید (Valproic acid) | مدل موشی ترانسژنیک AD (Transgenic mouse model of AD) | افزایش بیان H3 استیلهشده و کاهش اختلال حافظه و رسوب آمیلوئید-β | [141] |
| سودیم پروپیونات (Sodium propionate) | مدل موشی AD (AD mouse model) | بهبود اختلالات یادگیری و حافظه فضایی القاشده توسط Aβ1-42 با بازگرداندن پلاستیسیتی سیناپسی و کاهش نورواِینفلامیشن | [142] |
AD: Alzheimer’s disease; GPCR: G protein-coupled receptor; HDAC: Histone deacetylase; LPS: Lipopolysaccharide; ROS: Reactive oxygen species.
جدول 4. اثرات مفید اسیدهای چرب کوتاهزنجیر (SCFAs) یا داروهای مؤثر بر استیلدار شدن هیستونها (histone acetylation) در بیماری پارکینسون (Parkinson’s disease)
| SCFA یا دارو | نوع مطالعه | نتایج کلیدی | مرجع |
|---|---|---|---|
| سودیم بوتیرات (Sodium butyrate) | مدل موشی با علائم شبه PD القاشده توسط 6-OHDA (6-OHDA-induced PD-like symptoms in rats) | افزایش استیلاسیون هیستون H3 و سطح Bdnf و در نتیجه کاهش اختلالات حرکتی، نشانگرهای نورواِینفلامیشن و استرس اکسیداتیو | [143] |
| سودیم بوتیرات (Sodium butyrate) | موشهای PD القاشده با MPTP (Mice with MPTP-induced PD) | ارتقای سطح انتقالدهندههای عصبی استریاتال، بهبود عملکرد حرکتی و جلوگیری از مرگ نورونهای دوپامینرژیک از طریق بازگرداندن دیسبیوز میکروبی روده، کاهش اختلال سد رودهای و مهار مسیر TLR4/MyD88/NF-kB در روده و استریاتوم مغز | [144] |
| سودیم بوتیرات (Sodium butyrate) | سمیت القاشده با روتنون در سلولهای PC12 (Rotenone-induced toxicity in PC12 cells) | کاهش سمیت القاشده با روتنون از طریق افزایش H3K9ac و H3K27ac در ناحیه پروموتر PGC-1α و در نتیجه فعالسازی آوتوفاژی | [145] |
| سودیم بوتیرات (Sodium butyrate) | مدل موشی PD القاشده با α-synuclein (α-synuclein-induced rat model of PD) | افزایش دوپامین و استیلاسیون هیستون H3 و کاهش سایتوکاینهای پروالتهابی پس از درمان ترکیبی با سودیم بوتیرات و ترهالوز، یک القاکننده قوی آوتوفاژی، در موشهای α-synuclein پیشتکوینشده | [146] |
| سودیم بوتیرات (Sodium butyrate) | مدل موشی PD القاشده با 6-OHDA (6-OHDA–induced rat model of PD) | کاهش اختلالات شناختی در مرحله پراموتور در PD تجربی القاشده با 6-OHDA | [147] |
| سودیم بوتیرات (Sodium butyrate) | مدل Drosophila القاشده با روتنون برای PD (Rotenone-induced Drosophila model of PD) | بهبود اختلالات حرکتی | [148] |
| سودیم بوتیرات (Sodium butyrate) | مدل سلولی نورونهای دوپامینرژیک (A dopaminergic neuronal cell model) | افزایش سطح هیستون H3 استیله و افزایش بیان ژنهای مرتبط با ترمیم DNA | [149] |
| سودیم بوتیرات (Sodium butyrate) | موشهای PD القاشده با MPTP (MPTP-induced mice) | کاهش اختلال سد خونی مغزی (BBB) مرتبط با PD از طریق افزایش Occludin و ZO-1، افزایش Bcl-2 و کاهش Bax | [150] |
| سودیم بوتیرات (Sodium butyrate) | مدل موشی PD القاشده با رژیم غذایی پرچرب (A rat model of PD, high-fat diet-induced PD) | مکانیزم اپیژنتیک مرتبط با Fgf21 باعث اختلالات عملکردی در پاتولوژی PD مرتبط با مقاومت به انسولین شد و درمان ترکیبی با سودیم بوتیرات و متفورمین عملکرد حرکتی را از طریق افزایش Fgf21 بهبود داد | [151] |
| سودیم بوتیرات (Sodium butyrate) | مدل موشی PD القاشده با روتنون (Rotenone-induced PD mouse model) | کاهش دیسبیوز روده و بهبود عملکرد حرکتی؛ بازسازی ترکیب میکروبی روده و تنظیم متابولیسم SCFA در روده | [152] |
| سودیم بوتیرات (Sodium butyrate) | مدل موشی PD (A mouse model of PD) | کاهش شتاب غیرطبیعی α-synuclein، افزایش تیروزین هیدروکسیلاز، کاهش فعالسازی α-synuclein (>100%) در SNpc و مهار فعالسازی بیش از حد میکروگلیا | [153] |
| والپرویک اسید و سودیم بوتیرات (Valproic acid and sodium butyrate) | سمیت عصبی دوپامینرژیک القاشده با Mn در موشها (Mn-induced dopaminergic neurotoxicity in mice) | کاهش کاهش فعالیت حرکتی ناشی از Mn در موشها، احتمالاً از طریق مهار ژنهای Hdac | [153] |
| والپرویک اسید (Valproic acid) | موشهای PD با ضایعه 6-OHDA (6-OHDA-lesioned rats) | مهار سایتوکاینهای پروالتهابی و هیستون دهیدرالازها | [154] |
| والپرویک اسید (Valproic acid) | مدل موشی PD القاشده با روتنون (Rotenone rat model of PD) | کاهش فعالیت HDAC و افزایش استیلاسیون هیستون H3 در مغز موشهای تحت درمان ۴ هفتهای | [155] |
| والپرویک اسید (Valproic acid) | مدل موشی PD القاشده با MPTP (MPTP rat model of PD) | افزایش هیپر استیلاسیون H3K9 در سابستنتیا نیگرا موشهای FVBn و محافظت از نورونهای دوپامینرژیک | [156] |
| پروپیونات (Propionate) | مدل موشی PD القاشده با MPTP (MPTP rat model of PD) | کاهش اختلال عملکرد سد اپیتلیال روده مرتبط با PD از طریق مسیر سیگنالینگ AKT | [157] |
6-OHDA: 6-hydroxydopamine; BBB: Blood–brain barrier; HDAC: Histone deacetylase; IR: Insulin resistance; Mn: Manganese; MPTP: 1-methyl-4-phenyl-1,2,3,6-tetrathydropyridine; PD: Parkinson’s disease; SCFA: Short-chain fatty acid; SNpc: Substantia nigra pars compacta; ZO-1: Zonula occludens-1.
رژیم کتوژنیک و تغذیه با محدودیت زمانی در درمان آلزایمر و پارکینسون از طریق تغییرات اپیژنتیکی (Ketogenic diet & time-restricted feeding for the treatment of AD & PD via epigenetic changes)
اثرات بالقوه مفید رژیم کتوژنیک (ketogenic diet) و نقش محافظتی آن در برابر AD و PD با تنظیم میکروبیوم روده و متابولیتهای ثانویه آن (modulation of the gut microbiome and its secondary metabolites) و بهبود یکپارچگی نوروواسکولار (neurovascular integrity) از طریق مکانیزمهای اپیژنتیکی (epigenetic mechanisms) مرتبط است [154,155]. در مطالعهای توسط Yadav و همکارانش، رژیم غذایی انجمن قلب آمریکا (American Heart Association Diet) با رژیم کتوژنیک مدیترانهای اصلاحشده (modified Mediterranean ketogenic diet) بر تغییرات میکروبیوم روده و متابولیتهای ثانویه آن در سالمندان با اختلال شناختی خفیف (older adults with mild cognitive impairment) مقایسه شد [156]. نتایج نشان داد که رژیم کتوژنیک مدیترانهای اصلاحشده میتواند سطوح پروپیونات و بوتیرات (propionate and butyrate levels) و فراوانی خانوادههای باکتریایی Erysipelotriaceae, Christensenellaceae, Enterobacteriaceae, Akkermansia و Slackia را افزایش دهد، در حالی که فراوانی Bifidobacterium و Lachnobacterium کاهش یافت [156]. در مطالعهای دیگر، Ruan و همکارانش نشان دادند که مکانیزمهای اپیژنتیکی و ضدالتهابی رژیم کتوژنیک در مدل PD القا شده توسط لیپوپلیساکارید در موشها (lipopolysaccharide-induced rat PD model) مربوط به استیلدار شدن هیستون در ناحیه پروموتر mGluR5 و تنظیم مسیر سیگنالینگ Akt/GSK-3β/CREB است [157]. همچنین Cheng و همکارانش گزارش کردند که رژیم کتوژنیک قادر است سطح کتونهای خونی (blood ketone bodies) از جمله d-beta-hydroxybutyrate، استون (acetone) و استواستات (acetoacetate) را افزایش دهد که از طریق مکانیزمهای اپیژنتیکی (epigenetic mechanisms)، نورونهای دوپامینی منطقه سابستانتیا نیگرا (dopaminergic neurons of the substantia nigra) را در برابر نوروتوکسیسیتی 6-OHDA در مدل موشی PD (6-OHDA neurotoxicity in a rat model of PD) محافظت میکند، که این اثر از طریق افزایش گلوتاتیون (upregulating glutathione) صورت میگیرد [158].
تقریباً تمام بیماران AD (Alzheimer’s disease) تحت تأثیر اختلالات ریتم شبانهروزی (circadian disruptions) قرار دارند، و بنابراین مداخلات تنظیمکننده ریتم شبانهروزی (circadian-modulating interventions) میتوانند به عنوان استراتژیهای امیدوارکننده برای مهار بیان ژنهای مغزی غیرطبیعی وابسته به زمان روز و نورواِنتفلاماسیون (aberrant time-of-day brain transcription and neuroinflammation) در بیماران AD در نظر گرفته شوند. در این راستا، نتایج یک مطالعه اخیر نشان داد که تغذیه با محدودیت زمانی (time-restricted feeding, TRF; 7 ساعت در روز، پس از شروع فاز تاریک) در موشهای مسن (aged rats) قادر است میکروبهای روده مرتبط با تولید SCFAها (gut microbes associated with SCFA production) را تغییر دهد. تغییر از باکتریهای تولیدکننده استات به باکتریهای تولیدکننده بوتیرات (shift from acetate-producing toward butyrate-producing bacteria) توسط TRF اثرات مفیدی بر دیسبیوز روده و سلامت متابولیک (gut dysbiosis and metabolic health) دارد [159]. Desplats و همکارانش نیز کارایی رژیم TRF در موشهای مدل AD را بررسی کردند و دریافتند که TRF میتواند پاکسازی Aβ42 (Aβ42 clearance) را افزایش دهد، تجمع آمیلوئید (amyloid deposition) را کاهش دهد و الگوهای بیان روزانه چندین ژن مرتبط با نورواِنتفلاماسیون (daily expression patterns of multiple genes relevant to neuroinflammation) را با افزایش سطح β-hydroxybutyrate، یک تغییردهنده اپیژنتیکی (epigenetic modifier)، نرمال کند [160]. در مطالعهای دیگر، Shen و همکارانش نشان دادند که رژیم تقلید روزهداری (fasting mimicking diet; سه چرخه با مصرف 50٪ کالری روزانه استاندارد برای 1 روز، سپس 10٪ برای 2 روز و 4 روز تغذیه مجدد استاندارد) میتواند به حفظ عملکرد حرکتی (retention of motor function) و کاهش از دست رفتن نورونهای دوپامینی در سابستانتیا نیگرا (reducing the loss of dopaminergic neurons in the substantia nigra) در موشهای مدل PD القا شده با MPTP (1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine-induced PD model mice) کمک کند. این اثرات با افزایش فراوانی Firmicutes, Tenericutes و Opisthokonta، کاهش فراوانی Proteobacteria و سطوح بالاتر اسیدهای بوتیریک و والریک (butyric and valeric acids) همراه بود [161].
درمان پروبیوتیک و پریبیوتیک در آلزایمر و پارکینسون از طریق تغییرات اپیژنتیکی (Probiotic & prebiotic treatment of AD & PD via epigenetic changes)
پروبیوتیکها (Probiotics) به عنوان میکروارگانیسمهای زنده (living microorganisms) تعریف میشوند که در دوزهای مشخص، اثرات مفید سلامتی (health benefits) ایجاد میکنند. اگرچه مکانیزم دینامیک و زمینهای عملکرد پروبیوتیکها بر فیزیولوژی میزبان (host physiology) کاملاً مشخص نیست، اما ممکن است اثرات محافظتی خود را از طریق تغییرات اپیژنتیکی (epigenetic alterations) و تعدیل سیستم ایمنی میزبان (modulating the host immune system) اعمال کنند. پروبیوتیکها اثرات چندجانبه (pleiotropic effects) بر عملکرد سیستم عصبی مرکزی (central nervous system functions) از طریق محور میکروبیوم–روده–مغز (microbiota–gut–brain axis) دارند و اثرات نورحفاظتی (neuroprotective effects) آنها علیه AD و PD در چندین مطالعه گزارش شده است [162,163]. در یک مثال جالب، Choi و همکارانش اثرات نورحفاظتی یک ترکیب پروبیوتیک شامل Bifidobacterium animalis lactis, Lactobacillus rhamnosus GG و Lactobacillus acidophilus را در دو مدل موشی PD القا شده توسط روتنون (rotenone) یا MPTP بررسی کردند [164]. مشخص شد که این ترکیب پروبیوتیک میتواند سطح بوتیرات و فاکتورهای نوروتروفیک (neurotrophic factors) در مغز را افزایش دهد و سدیم بوتیرات (sodium butyrate) میتواند از دست رفتن نورونهای مسیر نیگروستریاتال القا شده توسط MPTP را مهار کند. در مطالعهای دیگر، درمان با Lactobacillus به همراه Bifidobacterium سبب افزایش سطح اسید بوتیریک (butyric acid) و استیلدار شدن Gsk3β در لیزین 15 (Gsk3β acetylation at lysine 15) شد که سطح فسفریلاسیون Tau را کاهش داد [165]. در مطالعهای توسط Tian و همکارانش، از Bifidobacterium breve CCFM1067 به صورت خوراکی در مدل موشی PD به مدت 5 هفته استفاده شد که نورونهای دوپامینی را با کاهش نورواِنتفلاماسیون و مهار هیپرفعالسازی سلولهای گلیال (glial cell hyperactivation) محافظت کرد [166]. تکمیل این پروبیوتیک همچنین تعداد باکتریهای مفید (Bifidobacterium و Akkermansia) را افزایش و فراوانی باکتریهای پاتوژنیک (Escherichia و Shigella) را در موشهای PD کاهش داد. نویسندگان همچنین اشاره کردند که فعالیت ضدالتهابی این پروبیوتیک در مغز یا روده مدل موشی PD القا شده توسط MPTP ممکن است به افزایش سطح SCFAها مرتبط باشد. Gaisford و همکارانش گزارش کردند که دوزدهی پروبیوتیک میتواند ترکیب باکتریایی میکروبیوم بیماران PD را در طول 48 ساعت تغییر دهد، از جمله تکثیر گونههای کومنسال تولیدکننده بوتیرات، افزایش سطح بوتیرات و در نتیجه کاهش التهاب و بهبود یکپارچگی اتصالات تنگ (tight junction integrity) [167]. در مطالعهای دیگر، درمان با Lactobacillus salivarius AP-32 و یک ترکیب پروبیوتیک/پریبیوتیک در موشهای PD القا شده توسط 6-OHDA موجب تغییرات در ترکیب میکروبیوتای روده، افزایش سطح اسید پروپیونیک و بوتیریک، و افزایش فعالیت آنزیمهای آنتیاکسیدان میزبان مانند کاتالاز (catalase)، گلوتاتیون پراکسیداز (glutathione peroxidase) و سوپراکسید دیسموتاز (superoxide dismutase) شد [168]. همچنین Mercer و همکارانش دریافتند که یک سوسپانسیون پروبیوتیک خوراکی (Symprove™) میتواند ترکیب میکروبیوتا را تغییر دهد، کاهش سطح بوتیرات را جلوگیری کند، نشانگرهای التهابی پلاسمایی را کاهش دهد و یکپارچگی روده را در مدل موشی PD در مراحل اولیه حفظ کند [169].
علاوه بر این، گزارش شده است که اثرات نورحفاظتی (neuroprotective effects) مکملهای پروبیوتیک علیه AD میتواند به افزایش سطح لاکتات و استات (lactate and acetate; an epigenetic modifier) در مغز موشهای AppNL-G-F مرتبط باشد [170]. Liu و همکارانش اثر پروبیوتیک Clostridium butyricum را علیه AD در موشهای ترنسژنیک APP/PS1 (APPswe/PS1dE9) بررسی کردند و دریافتند که درمان با C. butyricum میتواند اختلال شناختی، رسوبات Aβ و نورواِنتفلاماسیون میکروگلیایی (microglia-mediated neuroinflammation) را کاهش دهد. این اثرات از طریق تنظیم محور میکروبیوتا–روده–مغز (gut microbiota–gut–brain axis) mediated by butyrate اعمال میشوند [171].
علاوه بر پروبیوتیکهای (probiotics) ذکرشده در بالا، پریبیوتیکها (prebiotics) ــ یعنی مکملهای غذاییای که رشد باکتریهای مفید را تحریک میکنند ــ نیز دارای اثرات آنتاگونیستی بر بیماری آلزایمر (AD) هستند که احتمالاً از طریق مکانیسمهای اپیژنتیکی (epigenetic mechanisms) عمل میکنند. فروکتو-الیگوساکاریدها (fructo-oligosaccharides) ترکیبات تغذیهای غیرقابلهضم و از پریبیوتیکهای مهمی هستند که میکروبیوتای روده (gut microbiota) را تنظیم کرده و سطوح Aβ در مغز موشهای مدل آلزایمر را کاهش میدهند [172,173]. در مطالعهای، An و همکارانش گزارش کردند که بیان آنزیم Nep (یکی از آنزیمهای اصلی تجزیهکننده Aβ) در مغز موشهای مدل AD کاهش یافته است، اما درمان با فروکتو-الیگوساکاریدها موجب افزایش بیان Nep از طریق بهبود افزایش بیان Hdac2 در مغز این موشها شد [174].
اثرات پلیفنولها (polyphenol effects) بر AD و PD از طریق میکروبیوتای روده و تغییرات اپیژنتیکی
گیاهان بهدلیل داشتن پلیفنولها (polyphenols) به عنوان غذاهای عملکردی (functional foods) حیاتی در رژیم غذایی ما شناخته شدهاند. بر اساس ساختار شیمیایی متنوع، پلیفنولها به چهار خانواده اصلی شامل اسیدهای فنولی (phenolic acids)، فلاونوئیدها (flavonoids)، لیگنانها (lignans) و استیلبنها (stilbenes) تقسیم میشوند. چندین مطالعه in vivo نشان دادهاند که پلیفنولها قادرند AD و PD را با بازگرداندن تغییرات میکروبیوتای روده از طریق مکانیسمهای اپیژنتیکی (epigenetic mechanisms) بهبود بخشند. برای مثال، Qian و همکارانش اثرات محافظتی اسید کیکوریک (chicoric acid; CA)، یک اسید پلیفنولی استخراجشده از کاسنی و گل مخروطی بنفش (Echinacea purpurea)، را در موشهای مدل PD القا شده با MPTP بررسی کردند [175]. در این مطالعه نشان داده شد که CA قادر است ترکیب میکروبیوتای روده را تا حدی به حالت طبیعی بازگرداند، به طوری که فایلوم Firmicutes و جنسهای Lactobacillus و Ruminiclostridium افزایش یافته و فایلوم Bacteroidetes و جنس Parabacteroide کاهش یافتهاند. علاوه بر این، اثرات مفید CA در این مدل موشی PD با کاهش دیسبیوزیس میکروبی ناشی از MPTP از طریق تقویت انسجام اپیتلیوم کولون، بازگرداندن تولید طبیعی SCFAs و مهار مسیر سیگنالینگ TLR4/MyD88/NF-κB مرتبط بود. در مطالعهای دیگر، Zhanjun و همکارانش گزارش کردند که اسکوتلارین (scutellarin)، یک فلاونوئید استخراجشده از گیاه چینی Erigeron breviscapus، قادر است میکروبیوتای روده را تنظیم کرده و با مهار مسیر AMP–PKA–CREB–HDAC3 در میکروگلیا، باکتریهای مرتبط با التهاب را سرکوب کند در موشهای مدل آلزایمر (AD) [176]. همچنین، Wu و همکارانش نشان دادند که اثرات نورحفاظتی رزوراترول (resveratrol)، یک پلیفنول طبیعی با اثرات اپیژنتیکی مثبت قابل توجه، در حیوانات مدل PD القا شده با MPTP، با افزایش فراوانی Blautia، Erysipelotrichaceae، Prevotellaceae، Rikenellaceae و Alistipes، کاهش فراوانی Akkermansia و Lachnospiraceae و در نتیجه کاهش چندین سیتوکین التهابی مانند TNF-α، IL-6 و IL-1β همراه بود [177–179].
Zhang و همکارانش همچنین دریافتند که اثرات مفید پلیساکارید استخراجشده از قارچ کلم گل (Sparassis crispa; cauliflower mushroom) در AD با افزایش تکثیر جنسهای تولیدکننده بوتیرات (butyrate-producing genera) مانند Lachnospiraceae_UCG_010، Intestinaimonas، [Eubacterium] ventriosum group و Lachnospiraceae_UCG_001 و همچنین مهار تکثیر باکتریهای مرتبط با التهاب (مانند Escherichia/Shigella) مرتبط است [180]. سایر فیتوشیمیها (phytochemicals)، مانند آلیل سولفید (allyl sulfide)، یک ترکیب گوگردی ارگانیک موجود در سیر (garlic) نیز میتوانند اختلال مرتبط با سن در یکپارچگی سد روده (gut barrier integrity) را کاهش دهند و اختلالات میکروبیوتای مرتبط با سن و اختلالات حافظه را بهبود بخشند، از طریق کاهش سطح lncRNA غیرکدکننده Hotair در گردش خون و نرمالسازی H3K27ac در منطقه پروموتر ژن Ndnf در بافت هیپوکامپ موشهای مسن [181].
با توجه به اینکه فیتوشیمیها (phytochemicals) همچنین قادر به کاهش تغییرات اپیژنتیک ناشی از رادیکالهای آزاد اکسیژن (reactive oxygen species-mediated epigenetic alterations) هستند [182]، Nasuti و همکاران اثر آب الکترولیز شده و کاهشیافته (electrolyzed reduced water) بر میکروبیوتای مدفوع و نفوذپذیری روده (gut permeability) را در مدل موش صحرایی PD پس از قرار گرفتن بچهموشها در معرض آفتکش پرمترین (permethrin pesticide) بررسی کردند [183]. نتایج آنها نشان داد که Lachnospira در گروه مدل PD کمتر و Defluviitaleaceae بیشتر بود، و درمان با آب الکترولیز شده و کاهشیافته تأثیر مثبتی در بازگرداندن فراوانی Lachnospira و Defluviitaleaceae به مقادیر مشابه گروه کنترل داشت. آنها همچنین دریافتند که اثرات محافظتی آب الکترولیز شده و کاهشیافته در PD میتواند به افزایش تعداد باکتریهای تولیدکننده بوتیرات (butyrate-producing bacteria) مانند Blautia، گونههای ناشناخته (U.m.) از خانواده Lachnospiraceae و گونههای ناشناخته (U.m.) از خانواده Ruminococcaceae همراه با سطوح بالاتر اسید بوتیریک (butyric acid levels) نسبت داده شود.
علاوه بر فیتوشیمیها (phytochemicals)، مصرف فیبر (fiber intake) نیز با کاهش خطر ابتلا به AD (Alzheimer’s disease) در افراد مسن از طریق تغییرات اپیژنتیک مرتبط با میکروبیوم (microbiome-related epigenetic shifts) مرتبط دانسته شده است. برای مثال، Cuervo-Zanatta و همکاران دریافتند که مصرف فیبر محلول (soluble fiber) در موشهای نر ۶ ماهه APP/PS1 میتواند سطح بوتیرات (butyrate) را افزایش داده، سطح پروپیونات (propionate) را کاهش دهد، فعالسازی آستروسیتها (astrocyte activation) را بهبود بخشد و عملکرد شناختی (cognitive function) را ارتقا دهد [184]. قابل توجه است که درمان ترکیبی با آنتیبیوتیکهای غیرقابل جذب (nonabsorbable antibiotics) مانند وانکومایسین، پیمارسین، نئومایسین و باسیتراسین نیز به عنوان استراتژی امیدوارکنندهای برای کاهش آسیب سلولهای دوپامینرژیک ناشی از 6-OHDA و اختلالات حرکتی (motor impairment) مرتبط با آن مطرح شده است. این درمان اثرات خود را از طریق مدولاسیون میکروبیوم روده (gut microbiome modulation) و کاهش التهاب (alleviating inflammation) در مدل موش صحرایی با ضایعه یکطرفه PD اعمال میکند [185]. نویسندگان گزارش دادند که پس از درمان با آنتیبیوتیک، کاهش قابل توجهی در فراوانی فلاووباکتریها (Firmicutes) و افزایش فراوانی باکتریهای از شاخههای باکتروئیدتها (Bacteroidetes)، پروتئوباکتریا (Proteobacteria)، سیانوباکتریا (Cyanobacteria) و ورکو میکروبی (Verrucomicrobia) در موشهای تحت درمان با 6-OHDA مشاهده شد.
پیوند میکروبیوتای مدفوعی (Fecal Microbiota Transplantation; FMT) به عنوان یک استراتژی نوین (novel strategy) برای بهبود وضعیت بیماران مبتلا به AD و PD (Alzheimer’s disease and Parkinson’s disease) مطرح شده است. FMT به انتقال میکروبیوتای روده از یک اهداکننده به گیرنده (transfer of intestinal microbiota from one donor to a recipient)، معمولاً از طریق کولونوسکوپی (colonoscopy) گفته میشود [186]. با این حال، به دلیل نگرانیها در مورد ایمنی و اثربخشی بلندمدت (long-term safety and efficacy)، استفاده بالینی از FMT هنوز با چالش روبهرو است و در مراحل ابتدایی قرار دارد [187]. FMT میتواند یک رویکرد معتبر برای کاهش اختلالات نورودژنراتیو (neurodegenerative disorders) مانند AD و PD از طریق تغییرات اپیژنتیک (epigenetic changes) محسوب شود. در مطالعهای توسط Ye و همکاران، موشهای طبیعی یا موشهای PD-مانند القا شده با MPTP با FMT از موشهای سالم درمان شدند و نتایج نشان داد که FMT قادر به کاهش بیان α-synuclein، کاهش سطح SCFAهای مدفوعی (اسیدهای پروپیونیک، بوتیریک و n-والریک) و غیرفعالسازی مسیر TLR4/PI3K/AKT/NF-κB در موشهای PD بود [188]. در مطالعه دیگری، Liu و همکاران دریافتند که FMT میتواند پاتوژنز مشابه AD در موشهای ترنسژنیک APP/PS1 را کاهش دهد، با کاهش فسفریلاسیون تاو، کاهش سطوح Aβ40 و Aβ42 و افزایش پلاستیسیتی سیناپسی از طریق بازآرایی میکروبیوتای روده و افزایش سطح بوتیرات [189]. در خلاصه، شکل 2 اثرات تعاملی بین رژیم غذایی، میکروبیوتای روده و متابولیتهای آن را نشان میدهد که بر اپیژنوم (epigenome) اثر میگذارند تا التهاب کاهش یافته و تجمع پروتئین در AD و PD مهار شود.
شکل 2. تعامل بین رژیم غذایی، میکروبیوتای روده و متابولیتهای تولیدشده توسط روده که بر چشمانداز اپیژنتیک مرتبط با پاتوژنز AD و PD اثر میگذارند.
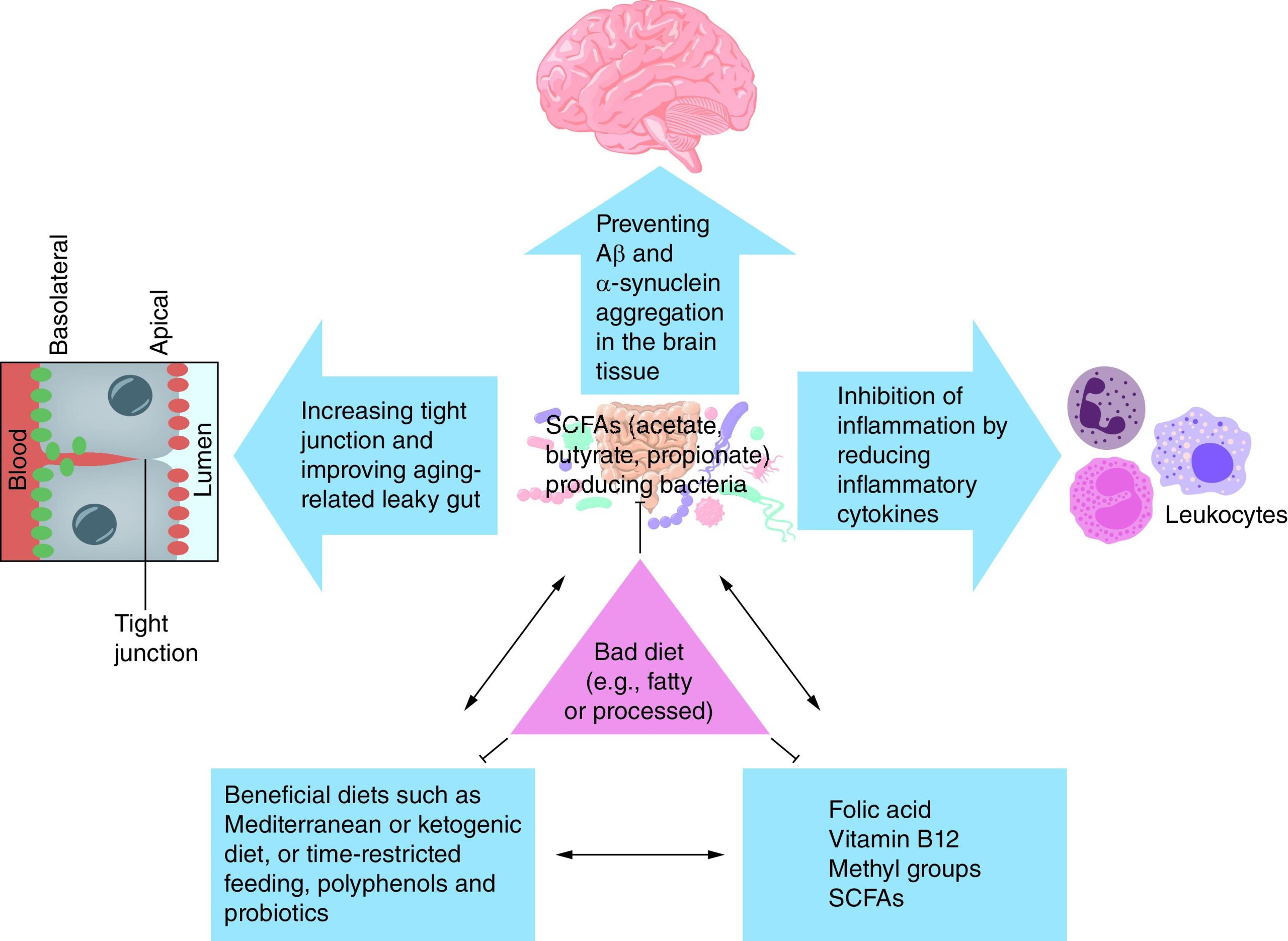
درمانهای غذایی (Dietary remedies) مانند رژیم کتوژنیک (ketogenic diet)، رژیم کمچرب (low-fat diet)، زمانبندی محدود غذا (time-restricted feeding)، پروبیوتیکها (probiotics) و پلیفنولها (polyphenols) به شدت بر ساختار و ترکیب میکروبیوتای روده (gut microbiota) و متابولیتهای آنها (metabolites) مانند پروپیونات، بوتیرات، استات (SCFAs)، فولات، ویتامین B12 و گروههای متیل برای واکنشهای متیلاسیون (methylation reactions) تأثیر میگذارند. افزایش سطح بوتیرات (butyrate) از طریق رژیمهای مفید مانند رژیم کتوژنیک، زمانبندی محدود غذا و پروبیوتیکها باعث مهار التهاب (suppress inflammation)، تقویت اتصالات محکم سلولهای رودهای (tight junctions) و کاهش leaky gut مرتبط با پیری میشود و در نتیجه از تجمع آمیلوئید-β و α-synuclein در بافت مغز جلوگیری میکند. رژیمهای مضر مانند رژیم پرچرب و غذاهای فرآوریشده (high-fat diet and processed foods) باعث کاهش سطح بوتیرات و سایر متابولیتهای ضروری و افزایش خطر توسعه AD و PD از طریق تغییرات اپیژنتیک، التهاب و تجمع پروتئین میشوند. علامتهای T-shape اطراف مثلث رژیم غذایی مضر نشاندهنده اثر توقف/مهاری (stalling/inhibitory effects) است.
نتیجهگیری
تحقیقات کنونی نشان میدهد که بین پاتوژنز AD و PD و تغییرات ترکیب میکروبیوتای روده (gut microbiome) همراه با تغییرات اپیژنتیک مرتبط با میکروبیوم (microbiome-related epigenetic changes) همبستگی قابل توجهی وجود دارد. شایان ذکر است که این تغییرات اپیژنتیک (epigenetic alterations) مرتبط با میکروبیوم میتوانند عملکرد مسیرهای نورو متابولیک و نورو شیمیایی (neurometabolic and neurochemical pathways) را در بافت مغز تغییر دهند، عمدتاً از طریق محور پیچیده ‘gut–brain’. بنابراین، این عوامل نقشهای محوری در افزایش حساسیت به ابتلا به AD و PD ایفا میکنند. در واقع، مکانیسمهای زیربنایی پاتوژنز AD و PD به هم تنیدهاند و شامل دیسبایوس روده (gut dysbiosis)، leaky gut، التهاب (inflammation)، اشتباه تاخوردگی و تجمع پروتئین (protein misfolding and aggregation) و تغییرات اپیژنتیک (epigenetic modifications) هستند. پژوهشگران به تدریج به درک ارتباط متقابل این عوامل دست یافتهاند که منجر به توسعه ابزارها و داروهای درمانی جدیدی شده است که هدف آنها بهبود علائم در مدلهای حیوانی AD و PD از طریق دستکاری میکروبیوم و بازبرنامهریزی اپیژنتیک مرتبط با میکروبیوم (microbiome-related epigenetic (re)programming) است. به طور امیدوارکننده، چندین مطالعه شواهد امیدوارکنندهای ارائه کردهاند که این مداخلات (interventions) میتوانند فنوتیپهای بیماری در انسانها را نیز بهبود دهند. این امر نشان میدهد که تحقیقات در حال حاضر ممکن است رویکردهای درمانی نوآورانه (novel therapeutic approaches) برای PD و AD ارائه کنند.
چشمانداز آینده
با وجود پیشرفتهای چشمگیر در درک مکانیسمهای دخیل در توسعه و پیشرفت AD و PD، پیچیدگی این اختلالات نورودژنراتیو (neurodegenerative disorders)، پتانسیل درمانی مؤثر در انسانها را محدود میکند. درک بهتر از ارتباط بین اجزای محور میکروبیوتا–روده–مغز (microbiota–gut–brain axis) میتواند مسیر را برای پایش پیشرفت AD و PD در مراحل مختلف هموار کرده و کارایی رویکردهای درمانی را افزایش دهد. با اینکه درمان AD و PD از طریق دستکاری میکروبیوم روده و بازبرنامهریزی اپیژنتیک مرتبط (epigenetic landscape) ممکن است روشهای درمانی نویدبخشی باشد، برخی مسائل علمی کلیدی و چالشهای فنی باید برای تسریع کاربردهای بالینی آنها مورد توجه قرار گیرد. انجام مطالعات بزرگمقیاس و چندملیتی (large-scale and multinational studies) با نمونههای بیشتر برای دستیابی به بینشهای عمیقتر در مورد تکرارپذیری و دقت درمانهای هدفمند میکروبیوم (microbiome-targeted therapies) ضروری است. علاوه بر این، برای بازتاب اثرات واقعی رژیمهای غذایی و پروبیوتیکهای خاص علیه AD و PD از طریق بازشکلدهی میکروبیوم روده و بازبرنامهریزی اپیژنتیک، استفاده از حیوانات بزرگتر مشابه انسان (مانند میمونها) یا مطالعات بالینی با نمونههای بیشتر باید انجام شود. تمرکز تلاشهای آینده باید بر بهینهسازی روشها برای درک پیچیدگی این بیماریها، ارتباط آنها با محور میکروبیوتا–روده–مغز و جلوگیری از نتایج متناقض ناشی از تفاوتهای جمعیتی، جغرافیایی، تکنولوژیکی و روششناسی باشد. در مجموع، این رویکردهای چندرشتهای (multidisciplinary approaches) چشمانداز امیدوارکنندهای برای ظهور راهبردهای درمانی نوین (novel therapeutic strategies) در سالهای آتی ارائه میکنند.
خلاصه اجرایی
تغییرات اپیژنتیک (Epigenetic modifications) و میکروبیوتای روده (Gut microbiota) در بیماریهای آلزایمر و پارکینسون
Alzheimer’s disease (AD) و Parkinson’s disease (PD) با اشتباه تا شدن پروتئینها (protein misfolding) و از دست رفتن عملکرد نورونها (loss of neuronal functions) مشخص میشوند که این آسیبها ناشی از جهشهای ژنتیکی (genetic mutations) و تغییرات اپیژنتیک (epigenetic alterations) هستند، جایی که میکروبیوتای روده (gut microbiota) نقش حیاتی در بینظمی اپیژنتیک ژنها و مسیرهای کلیدی مرتبط با این بیماریها (epigenetic dysregulation of key genes/pathways) ایفا میکند.
میکروبیوم روده (gut microbiome) شامل انواع متنوعی از میکروارگانیسمها مانند باکتریها، ویروسها، قارچها و پروتوزوآها (bacteria, viruses, fungi, and protozoa) است که تعداد آنها از سلولهای بدن انسان بیشتر است. تأثیر این میکروبیوم بر نفوذپذیری روده (intestinal permeability) میتواند باعث آزادسازی سیتوکینهای التهابی (inflammatory cytokines) شود که در نهایت به استرس اکسیداتیو (oxidative stress) و نوروفلاسیون (neuroinflammation) کمک میکند.
عوامل محیطی (environmental factors) مانند استفاده طولانیمدت از آنتیبیوتیکها، بیماریهای عفونی، مصرف غذاهای پرچرب و فرآوری شده (fatty and processed foods)، از طریق یک زنجیره متوالی از وقایع، نفوذپذیری مخاط روده (intestinal mucosa permeability) را افزایش داده و باکتریها و توکسینهای باکتریایی (bacterial toxins) را به جریان خون منتقل میکنند، که منجر به افزایش التهاب سیستمیک (systemic inflammation) و افزایش نفوذپذیری سد خونی–مغزی (BBB permeability) و در نتیجه افزایش حساسیت به AD و PD میشود.
بینظمیهای اپیژنتیک مرتبط با میکروبیوتای روده (gut microbiota-related epigenetic dysregulations) مسیرهایی مرتبط با پروتئینهای آمیلوئید (amyloid protein) و α-synuclein را تحت تأثیر قرار میدهند، و از این رو تغییرات اپیژنتیک ناشی از میکروبیوم و متابولیتهای آن (epigenetic modifications driven by the gut microbiome and its metabolites) میتوانند به عنوان هدفهای بالقوه درمانی برای AD و PD مطرح شوند.
رژیم غذایی و تغذیه محدود به زمان (Diet & Time-Restricted Feeding) برای درمان بیماریهای آلزایمر و پارکینسون از طریق تغییرات اپیژنتیک
تنظیم میکروبیوم روده (gut microbiome) و متابولیتهای آن از طریق روشهای غذایی مانند رژیم کتوژنیک (ketogenic diet) و تغذیه محدود به زمان (time-restricted feeding)، یک راهبرد درمانی مؤثر برای کاهش فرآیند پیری (mitigate the aging process) و کاهش خطر ابتلا به AD و PD محسوب میشود. این رویکرد بهویژه امیدبخش است، زیرا حدود ۶۰٪ از تغییرات میکروبیوم روده (variation in the gut microbiome) میتواند به رژیم غذایی (diet) نسبت داده شود.
نقش پلیفنولها (polyphenols) در بازسازی ترکیب باکتریهای روده و متابولیتهای آنها (reshaping the gut bacterial composition and their metabolites) میتواند با فراهم کردن زیرلایهها و تنظیمکنندههای آنزیمی برای تغییرات اپیژنتیک (substrates and enzymatic regulators for epigenetic modifications)، خطر ابتلا به AD و PD را کاهش دهد.
درمان پروبیوتیک (Probiotic) و پریبیوتیک (Prebiotic) در بیماری آلزایمر (AD) و پارکینسون (PD) از طریق تغییرات اپیژنتیک (Epigenetic changes)
تکمیل رژیم غذایی با پروبیوتیکها و پریبیوتیکها میتواند از بروز یا پیشرفت بیماری آلزایمر (AD) و پارکینسون (PD) که ناشی از اختلال در تعاملات میکروبیوتا–میزبان (Microbiota–host interplays) است، پیشگیری یا آن را کاهش دهد، از طریق تنظیم ترکیب میکروبیوتای روده (Gut microbiota composition) و در پی آن تغییر در ماشینآلات رونویسی (Transcriptional machinery) سلولهای مغزی (Brain cells).
پیوند میکروبیوتای مدفوع (Fecal microbiota transplantation) نیز یک استراتژی امیدوارکننده دیگر است که میتواند تأثیر تغییرات اپیژنتیک ناشی از التهاب (Inflammation-induced epigenetic alterations) را که در توسعه بیماری آلزایمر (AD) و پارکینسون (PD) نقش دارند، به طور مثبت تعدیل کند.
قدردانیها (Acknowledgments)
نویسندگان مایلاند از فریا اشرفی (Faria Ashrafi) صمیمانه تشکر کنند، بهخاطر مشارکتهای ارزشمند ایشان در تهیه و تولید تصاویر (Illustrations) این اثر.
پاورقیها (Footnotes)
مشارکت نویسندگان (Author contributions)
S. Nohesara پیشنویس (Drafted) مقاله را تهیه کرد. HM. Abdolmaleky مقاله را از منظر علمی بازبینی و اصلاح (Reviewed and revised) نمود. S. Thiagalingam و J-R. Zhou بازسازمانی (Reorganized) انجام دادند، و ویرایشهایی ارائه کردند و پروژه را به صورت مشترک هدایت کردند (Co-directed).
افشای وضعیت مالی (Financial disclosure)
S. Thiagalingam بخشی از حمایت مالی خود را از سوی مؤسسه ملی سلامت آمریکا (NIH, grant no. CA138509) دریافت کرد و فعالیتهای اولیه در آزمایشگاه نویسندگان با حمایت جایزه محقق مستقل NARSAD (NARSAD Independent Investigator Award) انجام شد. نویسندگان هیچ وابستگی یا مشارکت مالی دیگری با سازمان یا نهادی که دارای منافع مالی یا تضاد منافع (Financial conflict) مرتبط با موضوع یا مواد مطرح شده در مقاله باشد، ندارند، به جز مواردی که افشا شده است.
افشای تضاد منافع (Competing interests disclosure)
نویسندگان هیچ تضاد منافع (Competing interests) یا وابستگی مرتبط با سازمان یا نهادی که با موضوع یا مواد مطرح شده در مقاله مرتبط باشد، ندارند. این شامل اشتغال، مشاوره، حقالزحمه، مالکیت سهام یا اختیار سهام، شهادت تخصصی، دریافت یا در انتظار دریافت کمکهزینهها یا ثبت اختراع، یا حق امتیاز (Royalties) میشود.
افشای کمک در نگارش (Writing disclosure)
در تولید این مقاله هیچگونه کمک نگارشی (Writing assistance) استفاده نشده است.
کلیک کنید «References»
1.Bianchi VE, Herrera PF, Laura R. Effect of nutrition on neurodegenerative diseases. A systematic review. Nutr. Neurosci. 24(10), 810–834 (2021).
2.Vaquer-Alicea J, Diamond MI. Propagation of protein aggregation in neurodegenerative diseases. Annu. Rev. Biochem. 88, 785–810 (2019).
3.Vallerga CL, Zhang F, Fowdar J et al. Analysis of DNA methylation associates the cystine–glutamate antiporter SLC7A11 with risk of Parkinson’s disease. Nat. Commun. 11(1), 1238 (2020).
4.Xu Y, Xu L, Han M et al. Altered mitochondrial DNA methylation and mitochondrial DNA copy number in an APP/PS1 transgenic mouse model of Alzheimer disease. Biochem. Biophys. Res. Commun. 520(1), 41–46 (2019).
5.Andrade-Guerrero J, Santiago-Balmaseda A, Jeronimo-Aguilar P et al. Alzheimer’s disease: an updated overview of its genetics. Int. J. Mol. Sci. 24(4), 3754 (2023).
6.Nalls MA, Blauwendraat C, Vallerga CL et al. Identification of novel risk loci, causal insights, and heritable risk for Parkinson’s disease: a meta-analysis of genome-wide association studies. Lancet Neurol. 18(12), 1091–1102 (2019).
7.Zhou L-T, Liu D, Kang H-C et al. Tau pathology epigenetically remodels the neuron-glial cross-talk in Alzheimer’s disease. Sci. Adv. 9(16), eabq7105 (2023).
8.Zhang D, Zhang J, Wang Y et al. Targeting epigenetic modifications in Parkinson’s disease therapy. Med. Res. Rev. 43, 1748–1777 (2023).
9.Shen L, Wang C, Chen L, Wong G. Dysregulation of MicroRNAs and PIWI-Interacting RNAs in a Caenorhabditis elegans Parkinson’s disease model overexpressing human α-synuclein and influence of tdp-1. Front. Neurosci. 15, 600462 (2021).
10.Cammann D, Lu Y, Cummings MJ et al. Genetic correlations between Alzheimer’s disease and gut microbiome genera. Sci. Rep. 13(1), 5258 (2023).
11.Morais LH, Schreiber Iv HL, Mazmanian SK. The gut microbiota–brain axis in behaviour and brain disorders. Nat. Rev. Microbiol. 19(4), 241–255 (2021). ; •• Underlines microbiota–host interactions and the mechanisms implicating the gut microbiome in brain disorders via secretion of toxins, or health by short-chain fatty acids production, which regulates gut and blood–brain barrier permeability and numerous immune functions.
12.Zhu B, Wang X, Li L. Human gut microbiome: the second genome of human body. Protein Cell. 1(8), 718–725 (2010).
13.Zhao Y, Jaber V, Lukiw WJ. Gastrointestinal tract microbiome-derived pro-inflammatory neurotoxins in Alzheimer’s disease. J. Aging Sci. 9(Suppl. 5), 2 (2021). [PMC free article] [PubMed] [Google Scholar]
14.Zmora N, Suez J, Elinav E. You are what you eat: diet, health and the gut microbiota. Nat. Rev. Gastroenterol. Hepatol. 16(1), 35–56 (2019).
15.Ghosh TS, Shanahan F, O’toole PW. The gut microbiome as a modulator of healthy ageing. Nat. Rev. Gastroenterol. Hepatol. 19(9), 565–584 (2022).
16.Stan TL, Soylu-Kucharz R, Burleigh S et al. Increased intestinal permeability and gut dysbiosis in the R6/2 mouse model of Huntington’s disease. Sci. Rep. 10(1), 18270 (2020).
17.Sochocka M, Donskow-Łysoniewska K, Diniz BS, Kurpas D, Brzozowska E, Leszek J. The gut microbiome alterations and inflammation-driven pathogenesis of Alzheimer’s disease – a critical review. Mol. Neurobiol. 56, 1841–1851 (2019).
18.Kowalski K, Mulak A. Brain–gut–microbiota axis in Alzheimer’s disease. J. Neurogastroenterol. Motil. 25(1), 48 (2019).
19.Shandilya S, Kumar S, Jha NK, Kesari KK, Ruokolainen J. Interplay of gut microbiota and oxidative stress: perspective on neurodegeneration and neuroprotection. J. Adv. Res. 38, 223–244 (2022).
20.Tarawneh R, Holtzman DM. The clinical problem of symptomatic Alzheimer disease and mild cognitive impairment. Cold Spring Harb. Perspect. Med. 2(5), a006148 (2012).
21.Scheiblich H, Trombly M, Ramirez A, Heneka MT. Neuroimmune connections in aging and neurodegenerative diseases. Trends Immunol. 41(4), 300–312 (2020).
22.Power R, Prado-Cabrero A, Mulcahy R, Howard A, Nolan JM. The role of nutrition for the aging population: implications for cognition and Alzheimer’s disease. Annu. Rev. Food Sci. Technol. 10, 619–639 (2019).
23.Long JM, Holtzman DM. Alzheimer disease: an update on pathobiology and treatment strategies. Cell 179(2), 312–339 (2019).
24.Risacher SL, Anderson WH, Charil A et al. Alzheimer disease brain atrophy subtypes are associated with cognition and rate of decline. Neurology 89(21), 2176–2186 (2017).
25.Haass C, Kaether C, Thinakaran G, Sisodia S. Trafficking and proteolytic processing of APP. Cold Spring Harb. Perspect. Med. 2(5), a006270 2012).
26.Naj A, Schellenberg G, Alzheimer’s Disease Genetics Consortium (ADGC). Genomic variants, genes, and pathways of Alzheimer’s disease: an overview. Am. J. Med. Genet. B. Neuropsychiatr. Genet. 174, 5–26 (2017).
27.Wang Z, Zhao Y, Xu N et al. NEAT1 regulates neuroglial cell mediating Aβ clearance via the epigenetic regulation of endocytosis-related genes expression. Cell. Mol. Life Sci. 76, 3005–3018 (2019). ; • Demonstrates that epigenetic regulatory mechanisms are important in neuroglia-mediated Aβ clearance through regulation of Aβ endocytosis-related gene expression
28.Smith AR, Smith RG, Condliffe D et al. Increased DNA methylation near TREM2 is consistently seen in the superior temporal gyrus in Alzheimer’s disease brain. Neurobiol. Aging 47, 35–40 (2016).
29.Lardenoije R, Roubroeks JA, Pishva E et al. Alzheimer’s disease-associated (hydroxy) methylomic changes in the brain and blood. Clin. Epigenetics 11(1), 1–15 (2019).
30.Song H, Yang J, Yu W. Promoter hypomethylation of TGFBR3 as a risk factor of alzheimer’s disease: an integrated epigenomic-transcriptomic analysis. Front. cell dev. biol. 9, 825729 (2022).
31.Lang A-L, Eulalio T, Fox E et al. Methylation differences in Alzheimer’s disease neuropathologic change in the aged human brain. Acta Neuropathol. Commun. 10(1), 1–17 (2022).
32.Sommerer Y, Dobricic V, Schilling M et al. Entorhinal cortex epigenome-wide association study highlights four novel loci showing differential methylation in Alzheimer’s disease. Alzheimer’s Res. Ther. 15(1), 92 (2023).
33.Park H, Shin J, Kim Y, Saito T, Saido TC, Kim J. CRISPR/dCas9-Dnmt3a-mediated targeted DNA methylation of APP rescues brain pathology in a mouse model of Alzheimer’s disease. Transl. Neurodegener. 11(1), 1–12 (2022).
34.Santana DA, Smith MDaC, Chen ES. Histone modifications in alzheimer’s disease. Genes 14(2), 347 (2023).
35.Zotarelli-Filho IJ, Mogharbel BF, Irioda AC et al. State of the art of microRNAs signatures as biomarkers and therapeutic targets in Parkinson’s and Alzheimer’s diseases: a systematic review and meta-analysis. Biomedicines 11(4), 1113 (2023).
36.De Bastiani MA, Bellaver B, Brum WS et al. Hippocampal GFAP-positive astrocyte responses to amyloid and tau pathologies. Brain Behav. Immun. 110, 175–184 (2023).
37.Shireby G, Dempster EL, Policicchio S et al. DNA methylation signatures of Alzheimer’s disease neuropathology in the cortex are primarily driven by variation in non-neuronal cell-types. Nat. Commun. 13(1), 5620 (2022).
38.Kim E, Kim H, Jedrychowski MP et al. Irisin reduces amyloid-β by inducing the release of neprilysin from astrocytes following downregulation of ERK-STAT3 signaling. Neuron 111(22), 3619–3633.e8 (2023).
39.Nalivaeva NN, Belyaev ND, Turner AJ. New insights into epigenetic and pharmacological regulation of amyloid-degrading enzymes. Neurochem. Res. 41, 620–630 (2016).
40.Chen Y-A, Lu C-H, Ke C-C et al. Evaluation of class IIA histone deacetylases expression and in vivo epigenetic imaging in a transgenic mouse model of Alzheimer’s disease. Int. J. Mol. Sci. 22(16), 8633 (2021).
41.Hugais MM, Cobos SN, Bennett SA, Paredes J, Foran G, Torrente MP. Changes in histone H3 acetylation on lysine 9 accompany Aβ 1-40 overexpression in an Alzheimer’s disease yeast model. MicroPubl. Biol. 2021, (2021).
42.Blanco-Luquin I, Acha B, Urdánoz-Casado A et al. NXN gene epigenetic changes in an adult neurogenesis model of Alzheimer’s disease. Cells 11(7), 1069 (2022).
43.Zhang W, Young JI, Gomez L et al. Distinct CSF biomarker-associated DNA methylation in Alzheimer’s disease and cognitively normal subjects. Alzheimer’s Res. Ther. 15(1), 78 (2023).
44.Shao Y, Shaw M, Todd K et al. DNA methylation of TOMM40-APOE-APOC2 in Alzheimer’s disease. J. Hum. Genet. 63(4), 459–471 (2018).
45.Fertan E, Gendron WH, Wong AA, Hanson GM, Brown RE, Weaver IC. Noncanonical regulation of imprinted gene Igf2 by amyloid-beta 1–42 in Alzheimer’s disease. Sci. Rep. 13(1), 2043 (2023).
46.Klein H-U, Mccabe C, Gjoneska E et al. Epigenome-wide study uncovers large-scale changes in histone acetylation driven by tau pathology in aging and Alzheimer’s human brains. Nat. Neurosci. 22(1), 37–46 (2019).
47.Kakoty V, Kc S, Dubey SK, Yang C-H, Marathe SA, Taliyan R. Epigenetic regulation and autophagy modulation debilitates insulin resistance associated Alzheimer’s disease condition in rats. Metab. Brain Dis. 37(4), 927–944 (2022).
48.Maggiore A, Casale AM, Toscanelli W et al. Neuroprotective effects of PARP inhibitors in drosophila models of Alzheimer’s disease. Cells 11(8), 1284 (2022).
49.Bellver-Sanchis A, Singh Choudhary B, Companys-Alemany J et al. Structure-based virtual screening and in vitro and in vivo analyses revealed potent methyltransferase G9a inhibitors as prospective anti-Alzheimer’s agents. ChemMedChem 17(13), e202200002 (2022).
50.Liu H, Chu W, Gong L, Gao X, Wang W. MicroRNA-26b is upregulated in a double transgenic mouse model of Alzheimer’s disease and promotes the expression of amyloid-β by targeting insulin-like growth factor 1. Mol. Med. Rep. 13(3), 2809–2814 (2016).
51.Xing H, Guo S, Zhang Y, Zheng Z, Wang H. Upregulation of microRNA-206 enhances lipopolysaccharide-induced inflammation and release of amyloid-β by targeting insulin-like growth factor 1 in microglia. Mol. Med. Rep. 14(2), 1357–1364 (2016).
52.Geekiyanage H, Chan C. MicroRNA-137/181c regulates serine palmitoyltransferase and in turn amyloid β, novel targets in sporadic Alzheimer’s disease. J. Neurosci. 31(41), 14820–14830 (2011).
53.Fu L, Jiang G, Weng H, Dick GM, Chang Y, Kassab GS. Cerebrovascular miRNAs correlate with the clearance of Aβ through perivascular route in younger 3xTg-AD mice. Brain Pathol. 30(1), 92–105 (2020).
54.Bloem BR, Okun MS, Klein C. Parkinson’s disease. Lancet 397(10291), 2284–2303 (2021).
55.Lashuel HA, Overk CR, Oueslati A, Masliah E. The many faces of α-synuclein: from structure and toxicity to therapeutic target. Nat. Rev. Neurosci. 14(1), 38–48 (2013).
56.Nicoletti A, Luca A, Baschi R et al. Incidence of mild cognitive impairment and dementia in Parkinson’s disease: the Parkinson’s disease cognitive impairment study. Front. Aging Neurosci. 11, 21 (2019).
57.Dehay B, Bourdenx M, Gorry P et al. Targeting α-synuclein for treatment of Parkinson’s disease: mechanistic and therapeutic considerations. Lancet Neurol. 14(8), 855–866 (2015).
58.Li J, Jin M, Wang L, Qin B, Wang K. MDS clinical diagnostic criteria for Parkinson’s disease in China. J. Neurol. 264, 476–481 (2017).
59.Pajares M, Rojo AI, Manda G, Boscá L, Cuadrado A. Inflammation in Parkinson’s disease: mechanisms and therapeutic implications. Cells 9(7), 1687 (2020).
60.Li X, Li Q, Zhang Y et al. Nickel oxide nanoparticles increase α-synuclein amyloid formation and relevant overexpression of inflammatory mediators in microglia as a marker of Parkinson’s disease. Arab. J. Chem. 14(10), 103380 (2021).
61.Caputi V, Giron MC. Microbiome–gut–brain axis and toll-like receptors in Parkinson’s disease. Int. J. Mol. Sci. 19(6), 1689 (2018).
62.Perez-Pardo P, Dodiya HB, Engen PA et al. Role of TLR4 in the gut-brain axis in Parkinson’s disease: a translational study from men to mice. Gut 68(5), 829–843 (2019).
63.Kontopoulos E, Parvin JD, Feany MB. α-synuclein acts in the nucleus to inhibit histone acetylation and promote neurotoxicity. Hum. Mol. Genet. 15(20), 3012–3023 (2006).
64.Outeiro TF, Kontopoulos E, Altmann SM et al. Sirtuin 2 inhibitors rescue α-synuclein-mediated toxicity in models of Parkinson’s disease. Science 317(5837), 516–519 (2007).
65.Dai L, Wang J, He M et al. Lovastatin alleviates α-synuclein aggregation and phosphorylation in cellular models of synucleinopathy. Front. Mol. Neurosci. 14, 682320 (2021).
66.Mittal S, Bjørnevik K, Im DS et al. β2-Adrenoreceptor is a regulator of the α-synuclein gene driving risk of Parkinson’s disease. Science 357(6354), 891–898 (2017).
67.Harrison IF, Smith AD, Dexter DT. Pathological histone acetylation in Parkinson’s disease: neuroprotection and inhibition of microglial activation through SIRT 2 inhibition. Neurosci. Lett. 666, 48–57 (2018).
68.Musacchio T, Yin J, Kremer F et al. Temporal, spatial and molecular pattern of dopaminergic neurodegeneration in the AAV-A53T α-synuclein rat model of Parkinson’s disease. Behav. Brain Res. 432, 113968 (2022).
69.Toker L, Tran GT, Sundaresan J et al. Genome-wide histone acetylation analysis reveals altered transcriptional regulation in the Parkinson’s disease brain. Mol. Neurodegener. 16(1), 1–20 (2021).
70.Fedotova EY, Iakovenko EV, Abramycheva NY, Illarioshkin SN. SNCA gene methylation in Parkinson’s disease and multiple system atrophy. Epigenomes 7(1), 5 (2023).
71.Smith AR, Richards DM, Lunnon K, Schapira AH, Migdalska-Richards A. DNA methylation of α-synuclein intron 1 is significantly decreased in the frontal cortex of Parkinson’s individuals with GBA1 mutations. Int. J. Mol. Sci. 24(3), 2687 (2023).
72.Bakhit Y, Schmitt I, Hamed A et al. Methylation of alpha-synuclein in a Sudanese cohort. Parkinsonism Relat. Disord. 101, 6–8 (2022).
73.Henderson AR, Wang Q, Meechoovet B et al. DNA methylation and expression profiles of whole blood in Parkinson’s disease. Front. Genet. 12, 640266 (2021).
74.Wang R, Tong S, Wang M et al. CREB5 hypermethylation involved in the ganglioside GM1 therapy of Parkinson’s disease. Front. Aging Neurosci. 15, 1122647 (2023).
75.Magalingam KB, Somanath SD, Radhakrishnan AK. A glimpse into the genome-wide DNA methylation changes in 6-hydroxydopamine-induced in vitro model of Parkinson’s disease. Exp. Neurobiol. 32(3), 119 (2023).
76.Fu KA, Paul KC, Lu AT et al. DNA methylation-based surrogates of plasma proteins are associated with Parkinson’s disease risk. J. Neurol. Sci. 431, 120046 (2021).
77.Fisher DW, Tulloch J, Yu C-E, Tsuang D. A preliminary comparison of the methylome and transcriptome from the prefrontal cortex across Alzheimer’s disease and Lewy body dementia. J. Alzheimers Dis. Rep. 7(1), 279–297 (2023).
78.Gordevicius J, Li P, Marshall LL et al. Epigenetic inactivation of the autophagy–lysosomal system in appendix in Parkinson’s disease. Nat. Commun. 12(1), 5134 (2021).
79.Caldi Gomes L, Roser AE, Jain G et al. MicroRNAs from extracellular vesicles as a signature for Parkinson’s disease. Clin. Transl. Med. 11(4), e357 (2021).
80.Cappelletti C, Henriksen SP, Geut H et al. Transcriptomic profiling of Parkinson’s disease brains reveals disease stage specific expression changes. Acta Neuropathol. 146, 227–244 (2023).
81.Duan Y, Wang Y, Liu Y et al. Circular RNAs in Parkinson’s disease: reliable biological markers and targets for rehabilitation. Mol. Neurobiol. 60(6), 3261–3276 (2023).
82.Meng J, Wang F, Ji L et al. Comprehensive methylation profile of CSF cfDNA revealed pathogenesis and diagnostic markers for early-onset Parkinson’s disease. Epigenomics 13(20), 1637–1651 (2021).
83.Guhathakurta S, Kim J, Adams L et al. Targeted attenuation of elevated histone marks at SNCA alleviates α-synuclein in Parkinson’s disease. EMBO Mol. Med. 13(2), e12188 (2021).
84.Sardoiwala MN, Srivastava AK, Kaundal B, Karmakar S, Choudhury SR. Recuperative effect of metformin loaded polydopamine nanoformulation promoting EZH2 mediated proteasomal degradation of phospho-α-synuclein in Parkinson’s disease model. Nanomedicine 24, 102088 (2020).
85.Jowaed A, Schmitt I, Kaut O, Wüllner U. Methylation regulates alpha-synuclein expression and is decreased in Parkinson’s disease patients’ brains. J. Neurosci. 30(18), 6355–6359 (2010).
86.Srivastava AK, Choudhury SR, Karmakar S. Neuronal BMI-1 is critical for melatonin induced ubiquitination and proteasomal degradation of α-synuclein in experimental Parkinson’s disease models. Neuropharmacology 194, 108372 (2021).
87.Liu P, Sun L, Zhao X-L, Zhang P, Zhao X-M, Zhang J. PAR2-mediated epigenetic upregulation of α-synuclein contributes to the pathogenesis of Parkinson’s disease. Brain Res. 1565, 82–89 (2014).
88.Li B, Jiang Y, Xu Y, Li Y, Li B. Identification of miRNA-7 as a regulator of brain-derived neurotrophic factor/α-synuclein axis in atrazine-induced Parkinson’s disease by peripheral blood and brain microRNA profiling. Chemosphere 233, 542–548 (2019).
89.Xylaki M, Paiva I, Al-Azzani M et al. miR-101a-3p impairs synaptic plasticity and contributes to synucleinopathy. J. Parkinsons Dis. 13, 179–196 (2023).
90.Lin D, Zhang H, Zhang J et al. α-Synuclein induces neuroinflammation injury through the IL6ST-AS/STAT3/HIF-1α axis. Int. J. Mol. Sci. 24(2), 1436 (2023).
91.Schaffner SL, Wassouf Z, Hentrich T, Nuesch-Germano M, Kobor MS, Schulze-Hentrich JM. Distinct impacts of alpha-synuclein overexpression on the hippocampal epigenome of mice in standard and enriched environments. Neurobiol. Dis. 186, 106274 (2023).
92.Zhao A, Li Y, Niu M et al. SNCA hypomethylation in rapid eye movement sleep behavior disorder is a potential biomarker for Parkinson’s disease. J. Parkinsons Dis. 10(3), 1023–1031 (2020).
93.Kaut O, Kuchelmeister K, Moehl C, Wüllner U. 5-methylcytosine and 5-hydroxymethylcytosine in brains of patients with multiple system atrophy and patients with Parkinson’s disease. J. Chem. Neuroanat. 96, 41–48 (2019).
94.Zhou T, Lin D, Chen Y et al. α-synuclein accumulation in SH-SY5Y cell impairs autophagy in microglia by exosomes overloading miR-19a-3p. Epigenomics 11(15), 1661–1677 (2019).
95.Tao H, Liu Y, Hou Y. miRNA-384-5p regulates the progression of Parkinson’s disease by targeting SIRT1 in mice and SH-SY5Y cell. Int. J. Mol. Med. 45(2), 441–450 (2020).
96.Obrenovich ME. Leaky gut, leaky brain? Microorganisms 6(4), 107 (2018). ; •• Explores the importance of leaky gut and microbiota dysbiosis in altering blood–brain barrier permeability and increasing the risk of neurological disorders such as Alzheimer’s disease or Parkinson’s disease.
97.Hollander D, Kaunitz JD. The “leaky gut”: tight junctions but loose associations? Dig. Dis. Sci. 65(5), 1277–1287 (2020).
98.Nagpal R, Mainali R, Ahmadi S et al. Gut microbiome and aging: physiological and mechanistic insights. Nutr. Healthy Aging 4(4), 267–285 (2018).
99.Qian Y, Yang X, Xu S et al. Detection of microbial 16S rRNA gene in the blood of patients with Parkinson’s disease. Front. Aging Neurosci. 10, 156 (2018).
100.Forsyth CB, Shannon KM, Kordower JH et al. Increased intestinal permeability correlates with sigmoid mucosa alpha-synuclein staining and endotoxin exposure markers in early Parkinson’s disease. PLOS ONE 6(12), e28032 (2011).
101.Park A-M, Tsunoda I. Helicobacter pylori infection in the stomach induces neuroinflammation: the potential roles of bacterial outer membrane vesicles in an animal model of Alzheimer’s disease. Inflamm. Regen. 42(1), 39 (2022).
102.Wang S, Prajapati SK, Mishra SP, Jain S, Yadav H. Protection of cognitive decline and Alzheimer’s disease progression by a human origin-probiotic biotherapy. Alzheimers Dement. 18, e066137 (2022).
103.Li H, Liu C-C, Zheng H, Huang TY. Amyloid, tau, pathogen infection and antimicrobial protection in Alzheimer’s disease–conformist, nonconformist, and realistic prospects for AD pathogenesis. Transl. Neurodegener. 7, 1–16 (2018).
104.Shukla PK, Delotterie DF, Xiao J et al. Alterations in the gut–microbial–inflammasome–brain axis in a mouse model of Alzheimer’s disease. Cells 10(4), 779 (2021). ; • Proposes a model wherein modulating the gut microbiota and targeting gut–microbial–inflammasome components represents a promising strategy for preventing or combating Alzheimer’s disease-related neurological disorders in genetically susceptible individuals.
105.Kaya-Tilki E, Dikmen M. Neuroprotective effects of some epigenetic modifying drugs’ on Chlamydia pneumoniae-induced neuroinflammation: a novel model. PLOS ONE 16(11), e0260633 (2021).
106.Ahmadi S, Wang S, Nagpal R et al. A human-origin probiotic cocktail ameliorates aging-related leaky gut and inflammation via modulating the microbiota/taurine/tight junction axis. JCI Insight 5(9), e132055 (2020). ; •• Interesting study showing a human-origin probiotic cocktail ameliorates aging-related leaky gut and inflammation by increasing the levels of epigenetic modifiers (butyrateand propionate) and hence improves tight junctions.
107.Ren M, Li H, Fu Z, Li Q. Centenarian-sourced lactobacillus casei combined with dietary fiber complex ameliorates brain and gut function in aged mice. Nutrients 14(2), 324 (2022).
108.Zhao X, Kong M, Wang Y et al. Nicotinamide mononucleotide improves the Alzheimer’s disease by regulating intestinal microbiota. Biochem. Biophys. Res. Commun. 670, 27–35 (2023).
109.Unger MM, Spiegel J, Dillmann K-U et al. Short chain fatty acids and gut microbiota differ between patients with Parkinson’s disease and age-matched controls. Parkinsonism Relat. Disord. 32, 66–72 (2016). ; • Demonstrating the importance of gut microbiota and their metabolites as epigenetic modifiers in the quality of life, health and prevention of Parkinson’s disease.
110.Vogt NM, Romano KA, Darst BF et al. The gut microbiota-derived metabolite trimethylamine N-oxide is elevated in Alzheimer’s disease. Alzheimer’s Res. Ther. 10, 1–8 (2018).
111.Choudhury SP, Bano S, Sen S et al. Altered neural cell junctions and ion-channels leading to disrupted neuron communication in Parkinson’s disease. NPJ Parkinson’s Dis. 8(1), 66 (2022).
112.Sampson TR, Debelius JW, Thron T et al. Gut microbiota regulate motor deficits and neuroinflammation in a model of Parkinson’s disease. Cell 167(6), 1469–1480. e1412 (2016).
113.Guo X, Tang P, Hou C et al. Integrated microbiome and host transcriptome profiles link Parkinson’s disease to Blautia genus: evidence from feces, blood, and brain. Front. Microbiol. 13, 875101 (2022).
114.Ferreiro AL, Choi J, Ryou J et al. Gut microbiome composition may be an indicator of preclinical Alzheimer’s disease. Sci. Transl. Med. 15(700), eabo2984 (2023).
115.Kundu P, Torres ERS, Stagaman K et al. Integrated analysis of behavioral, epigenetic, and gut microbiome analyses in App NL-GF, App NL-F, and wild type mice. Sci. Rep. 11(1), 4678 (2021).
116.Blacher E, Levy M, Tatirovsky E, Elinav E. Microbiome-modulated metabolites at the interface of host immunity. J. Immunol. 198(2), 572–580 (2017).
117.Chen S-J, Chen C-C, Liao H-Y et al. Association of fecal and plasma levels of short-chain fatty acids with gut microbiota and clinical severity in patients with Parkinson disease. Neurology 98(8), e848–e858 (2022). ; • A relatively large human study showing that plasma and fecal short-chain fatty acids (such as butyrate) levels are linked to disease severity in patients with Parkinson’s disease.
118.Dalile B, Van Oudenhove L, Vervliet B, Verbeke K. The role of short-chain fatty acids in microbiota–gut–brain communication. Nat. Rev. Gastroenterol. Hepatol. 16(8), 461–478 (2019).
119.Zhang L, Wang Y, Xiayu X et al. Altered gut microbiota in a mouse model of Alzheimer’s disease. J. Alzheimer’s Dis. 60(4), 1241–1257 (2017).
120.Marizzoni M, Cattaneo A, Mirabelli P et al. Short-chain fatty acids and lipopolysaccharide as mediators between gut dysbiosis and amyloid pathology in Alzheimer’s disease. J. Alzheimer’s Dis. 78(2), 683–697 (2020).
121.Xu Y, Wei S, Zhu L et al. Low expression of the intestinal metabolite butyric acid and the corresponding memory pattern regulate HDAC4 to promote apoptosis in rat hippocampal neurons. Ecotoxicol. Environ. Saf. 253, 114660 (2023).
122.Sarkar S, Abujamra AL, Loew JE, Forman LW, Perrine SP, Faller DV. Histone deacetylase inhibitors reverse CpG methylation by regulating DNMT1 through ERK signaling. Anticancer Res. 31(9), 2723–2732 (2011). [PubMed] [Google Scholar]
123.Xie A, Ensink E, Li P et al. Bacterial butyrate in Parkinson’s disease is linked to epigenetic changes and depressive symptoms. Mov. Disord. 37(8), 1644–1653 (2022).
124.Sun Y, Zhang H, Zhang X et al. Promotion of astrocyte–neuron glutamate–glutamine shuttle by SCFA contributes to the alleviation of Alzheimer’s disease. Redox. Biol. 62, 102690 (2023).
125.Avagliano C, Coretti L, Lama A et al. Dual-hit model of Parkinson’s disease: impact of dysbiosis on 6-Hydroxydopamine-insulted mice – neuroprotective and anti-inflammatory effects of butyrate. Int. J. Mol. Sci. 23(12), 6367 (2022).
126.Chen G, Ran X, Li B et al. Sodium butyrate inhibits inflammation and maintains epithelium barrier integrity in a TNBS-induced inflammatory bowel disease mice model. EBioMedicine 30, 317–325 (2018).
127.Getachew B, Csoka AB, Bhatti A, Copeland RL, Tizabi Y. Butyrate protects against salsolinol-induced toxicity in SH-SY5Y cells: implication for Parkinson’s disease. Neurotox. Res. 38, 596–602 (2020).
128.Fernando W, Martins IJ, Morici M et al. Sodium butyrate reduces brain amyloid-β levels and improves cognitive memory performance in an Alzheimer’s disease transgenic mouse model at an early disease stage. J. Alzheimer’s Dis. 74(1), 91–99 (2020).
129.Bayazid AB, Jeong YH, Jeong SA, Lim BO. Sodium butyrate alleviates potential Alzheimer’s disease in vitro by suppressing Aβ and tau activation and ameliorates Aβ-induced toxicity. Food Agric. Immunol. 34(1), 2234100 (2023). [Google Scholar]
130.Jiang Y, Li K, Li X, Xu L, Yang Z. Sodium butyrate ameliorates the impairment of synaptic plasticity by inhibiting the neuroinflammation in 5XFAD mice. Chem. Biol. Interact. 341, 109452 (2021).
131.Govindarajan N, Agis-Balboa RC, Walter J, Sananbenesi F, Fischer A. Sodium butyrate improves memory function in an Alzheimer’s disease mouse model when administered at an advanced stage of disease progression. J. Alzheimer’s Dis. 26(1), 187–197 (2011).
132.Wang C, Zheng D, Weng F, Jin Y, He L. Sodium butyrate ameliorates the cognitive impairment of Alzheimer’s disease by regulating the metabolism of astrocytes. Psychopharmacology (Berl.) 239,215–227 (2022).
133.Kilgore M, Miller CA, Fass DM et al. Inhibitors of class 1 histone deacetylases reverse contextual memory deficits in a mouse model of Alzheimer’s disease. Neuropsychopharmacology 35(4), 870–880 (2010).
134.Nuutinen T, Suuronen T, Kauppinen A, Salminen A. Valproic acid stimulates clusterin expression in human astrocytes: Implications for Alzheimer’s disease. Neurosci. Lett. 475(2), 64–68 (2010).
135.Yuan B, Liu M, Gong Y et al. Sodium butyrate exerts antioxidant stress effects and attenuates Aβ25-35-induced cytotoxicity in PC12 cells. Arch. Biochem. Biophys. 731, 109448 (2022).
136.Sun J, Yuan B, Wu Y et al. Sodium butyrate protects N2a cells against Aβ toxicity in vitro. Mediators Inflamm. 2020, 7605160 (2020).
137.Ho L, Ono K, Tsuji M, Mazzola P, Singh R, Pasinetti GM. Protective roles of intestinal microbiota derived short chain fatty acids in Alzheimer’s disease-type beta-amyloid neuropathological mechanisms. Expert Rev. Neurother. 18(1), 83–90 (2018).
138.Long ZM, Zhao L, Jiang R et al. Valproic acid modifies synaptic structure and accelerates neurite outgrowth via the glycogen synthase kinase-3β signaling pathway in an Alzheimer’s disease model. CNS Neurosci. Ther. 21(11), 887–897 (2015).
139.Noh H, Seo H. Age-dependent effects of valproic acid in Alzheimer’s disease (AD) mice are associated with nerve growth factor (NGF) regulation. Neuroscience 266, 255–265 (2014).
140.Sorial ME, El Sayed NSED. Protective effect of valproic acid in streptozotocin-induced sporadic Alzheimer’s disease mouse model: possible involvement of the cholinergic system. Naunyn. Schmiedebergs Arch. Pharmacol. 390, 581–593 (2017).
141.Xuan A-G, Pan X-B, Wei P et al. Valproic acid alleviates memory deficits and attenuates amyloid-β deposition in transgenic mouse model of Alzheimer’s disease. Mol. Neurobiol. 51, 300–312 (2015).
142.Lang W, Li X, Wang Y et al. Sodium propionate improves cognitive and memory function in mouse models of Alzheimer’s disease. Neurosci. Lett. 791, 136887 (2022).
143.Sharma S, Taliyan R, Singh S. Beneficial effects of sodium butyrate in 6-OHDA induced neurotoxicity and behavioral abnormalities: modulation of histone deacetylase activity. Behav. Brain Res. 291, 306–314 (2015).
144.Guo T-T, Zhang Z, Sun Y et al. Neuroprotective effects of sodium butyrate by restoring gut microbiota and inhibiting TLR4 signaling in mice with MPTP-induced Parkinson’s disease. Nutrients 15(4), 930 (2023).
145.Zhang Y, Xu S, Qian Y et al. Sodium butyrate attenuates rotenone-induced toxicity by activation of autophagy through epigenetically regulating PGC-1α expression in PC12 cells. Brain Res. 1776, 147749 (2022).
146.Kakoty V, Kc S, Dubey SK, Yang C-H, Taliyan R. Neuroprotective effects of trehalose and sodium butyrate on preformed fibrillar form of α-synuclein-induced rat model of Parkinson’s disease. ACS Chem. Neurosci. 12(14), 2643–2660 (2021).
147.Rane P, Shields J, Heffernan M, Guo Y, Akbarian S, King JA. The histone deacetylase inhibitor, sodium butyrate, alleviates cognitive deficits in pre-motor stage PD. Neuropharmacology 62(7), 2409–2412 (2012).
148.Laurent RS, O’brien LM, Ahmad S. Sodium butyrate improves locomotor impairment and early mortality in a rotenone-induced Drosophila model of Parkinson’s disease. Neuroscience 246, 382–390 (2013).
149.Paiva I, Pinho R, Pavlou MA et al. Sodium butyrate rescues dopaminergic cells from alpha-synuclein-induced transcriptional deregulation and DNA damage. Hum. Mol. Genet. 26(12), 2231–2246 (2017).
150.Liu J, Wang F, Liu S et al. Sodium butyrate exerts protective effect against Parkinson’s disease in mice via stimulation of glucagon like peptide-1. J. Neurol. Sci. 381, 176–181 (2017).
151.Kakoty V, Kc S, Yang C-H, Dubey SK, Taliyan R. Exploring the epigenetic regulated modulation of fibroblast growth factor 21 involvement in high-fat diet associated parkinson’s disease in rats. ACS Chem. Neurosci. 14(4), 725–740 (2023).
152.Zhang Y, Xu S, Qian Y et al. Sodium butyrate ameliorates gut dysfunction and motor deficits in a mouse model of Parkinson’s disease by regulating gut microbiota. Front. Aging Neurosci. 15, 1099018 (2023).
153.Hou Y, Li X, Liu C et al. Neuroprotective effects of short-chain fatty acids in MPTP induced mice model of Parkinson’s disease. Exp. Gerontol. 150, 111376 (2021).
154.Dilmore AH, Martino C, Neth BJ et al. Effects of a ketogenic and low-fat diet on the human metabolome, microbiome, and foodome in adults at risk for Alzheimer’s disease. Alzheimers. Dement. 19(11), 4805–4816 (2023).
155.Ma D, Wang AC, Parikh I et al. Ketogenic diet enhances neurovascular function with altered gut microbiome in young healthy mice. Sci. Rep. 8(1), 6670 (2018).
156.Nagpal R, Neth BJ, Wang S, Craft S, Yadav H. Modified Mediterranean–ketogenic diet modulates gut microbiome and short-chain fatty acids in association with Alzheimer’s disease markers in subjects with mild cognitive impairment. EBioMedicine 47, 529–542 (2019).
157.Zhu Y, Tang X, Cheng Z, Dong Q, Ruan G. The anti-inflammatory effect of preventive intervention with ketogenic diet mediated by the histone acetylation of mGluR5 promotor region in rat Parkinson’s disease model: a dual-tracer PET study. Parkinsons Dis. 2022, 3506213 (2022).
158.Cheng B, Yang X, An L, Gao B, Liu X, Liu S. Ketogenic diet protects dopaminergic neurons against 6-OHDA neurotoxicity via up-regulating glutathione in a rat model of Parkinson’s disease. Brain Res. 1286, 25–31 (2009).
159.Hernandez AR, Kemp KM, Burke SN, Buford TW, Carter CS. Influence of aging, macronutrient composition and time-restricted feeding on the Fischer344 x brown norway rat gut microbiota. Nutrients 14(9), 1758 (2022).
160.Whittaker DS, Akhmetova L, Carlin D et al. Circadian modulation by time-restricted feeding rescues brain pathology and improves memory in mouse models of Alzheimer’s disease. Cell Metab. 35(10), 1704–1721.e6 (2023). ; •• An interesting study suggesting that dietary programming, such as time-restricted feeding, may serve as effective strategies for enhancing memory in individuals with Alzheimer’s disease.
161.Zhou Z-L, Jia X-B, Sun M-F et al. Neuroprotection of fasting mimicking diet on MPTP-induced Parkinson’s disease mice via gut microbiota and metabolites. Neurotherapeutics 16, 741–760 (2019).
162.Hsieh T-H, Kuo C-W, Hsieh K-H et al. Probiotics alleviate the progressive deterioration of motor functions in a mouse model of Parkinson’s disease. Brain Sci. 10(4), 206 (2020).
163.Asl ZR, Sepehri G, Salami M. Probiotic treatment improves the impaired spatial cognitive performance and restores synaptic plasticity in an animal model of Alzheimer’s disease. Behav. Brain Res. 376, 112183 (2019).
164.Srivastav S, Neupane S, Bhurtel S et al. Probiotics mixture increases butyrate, and subsequently rescues the nigral dopaminergic neurons from MPTP and rotenone-induced neurotoxicity. J. Nutr. Biochem. 69, 73–86 (2019).
165.Zhang Y, Shen Y, Liufu N et al. Transmission of Alzheimer’s disease-associated microbiota dysbiosis and its impact on cognitive function: evidence from mouse models and human patients. Mol. Psychiatry (2023).
166.Li T, Chu C, Yu L et al. Neuroprotective effects of Bifidobacterium breve CCFM1067 in MPTP-induced mouse models of Parkinson’s disease. Nutrients 14(21), 4678 (2022).
167.Ghyselinck J, Verstrepen L, Moens F et al. Influence of probiotic bacteria on gut microbiota composition and gut wall function in an in-vitro model in patients with Parkinson’s disease. Int. J. Pharm: X 3, 100087 (2021).
168.Tsao S-P, Nurrahma BA, Kumar R et al. Probiotic enhancement of antioxidant capacity and alterations of gut microbiota composition in 6-hydroxydopamin-induced parkinson’s disease rats. Antioxidants 10(11), 1823 (2021).
169.Sancandi M, De Caro C, Cypaite N et al. Effects of a probiotic suspension Symprove™ on a rat early-stage Parkinson’s disease model. Front. Aging Neurosci. 14, 1525 (2023).
170.Kaur H, Golovko S, Golovko MY, Singh S, Darland DC, Combs CK. Effects of probiotic supplementation on short chain fatty acids in the App NL-GF mouse model of Alzheimer’s disease. J. Alzheimer’s Dis. 76(3), 1083–1102 (2020).
171.Sun J, Xu J, Yang B et al. Effect of Clostridium butyricum against microglia-mediated neuroinflammation in Alzheimer’s disease via regulating gut microbiota and metabolites butyrate. Mol. Nutr. Food Res. 64(2), 1900636 (2020).
172.Yen C-H, Wang C-H, Wu W-T, Chen H-L. Fructo-oligosaccharide improved brain β-amyloid, β-secretase, cognitive function, and plasma antioxidant levels in D-galactose-treated Balb/cJ mice. Nutr. Neurosci. 20(4), 228–237 (2017).
173.Sun J, Liu S, Ling Z et al. Fructooligosaccharides ameliorating cognitive deficits and neurodegeneration in APP/PS1 transgenic mice through modulating gut microbiota. J. Agric. Food Chem. 67(10), 3006–3017 (2019).
174.Wu S, Zhang J, Jiang C, Wang S, Que R, An L. Up-regulation of neprilysin mediates the protection of fructo-oligosaccharides against Alzheimer’s disease. Food Funct. 11(7), 6565–6572 (2020).
175.Wang N, Feng B-N, Hu B, Cheng Y-L, Guo Y-H, Qian H. Neuroprotection of chicoric acid in a mouse model of Parkinson’s disease involves gut microbiota and TLR4 signaling pathway. Food Funct. 13(4), 2019–2032 (2022).
176.Zhang S, Wei D, Lv S et al. Scutellarin modulates the microbiota–gut–brain axis and improves cognitive impairment in APP/PS1 mice. J. Alzheimer’s Dis. 89(3), 955–975 (2022).
177.Pyo IS, Yun S, Yoon YE, Choi J-W, Lee S-J. Mechanisms of aging and the preventive effects of resveratrol on age-related diseases. Molecules 25(20), 4649 (2020).
178.Grinan-Ferre C, Bellver-Sanchis A, Izquierdo V et al. The pleiotropic neuroprotective effects of resveratrol in cognitive decline and Alzheimer’s disease pathology: from antioxidant to epigenetic therapy. Ageing Res. Rev. 67, 101271 (2021).
179.Tao J, An Y, Xu L et al. The protective role of microbiota in the prevention of MPTP/P-induced Parkinson’s disease by resveratrol. Food Funct. 14(10), 4647–4661 (2023).
180.Zhang W, Guo Y, Cheng Y, Yao W, Qian H. Neuroprotective effects of polysaccharide from Sparassis crispa on Alzheimer’s disease-like mice: involvement of microbiota–gut–brain axis. Int. J. Biol. Macromol. 225, 974–986 (2023).
181.Behera J, Kelly KE, Tyagi N. Altered non-coding RNA-histone acetylation regulatory circuit is associated with cognitive impairment via gut dysbiosis in aging mice. FASEB J. 33(S1), 714.713 (2019). [Google Scholar]
182.Abdolmaleky HM, Zhou J-R. Underlying mechanisms of brain aging and neurodegenerative diseases as potential targets for preventive or therapeutic strategies using phytochemicals. Nutrients 15(15), 3456 (2023).
183.Bordoni L, Gabbianelli R, Fedeli D et al. Positive effect of an electrolyzed reduced water on gut permeability, fecal microbiota and liver in an animal model of Parkinson’s disease. PLOS ONE 14(10), e0223238 (2019).
184.Cuervo-Zanatta D, Syeda T, Sánchez-Valle V et al. Dietary fiber modulates the release of gut bacterial products preventing cognitive decline in an Alzheimer’s mouse model. Cell. Mol. Neurobiol. 43(4), 1595–1618 (2023).
185.Koutzoumis DN, Vergara M, Pino J et al. Alterations of the gut microbiota with antibiotics protects dopamine neuron loss and improve motor deficits in a pharmacological rodent model of Parkinson’s disease. Exp. Neurol. 325, 113159 (2020).
186.Bakker GJ, Nieuwdorp M. Fecal microbiota transplantation: therapeutic potential for a multitude of diseases beyond Clostridium difficile. Microbiol. Spectr. 5(4), (2017).
187.Wortelboer K, Nieuwdorp M, Herrema H. Fecal microbiota transplantation beyond Clostridioides difficile infections. EBioMedicine 44, 716–729 (2019).
188.Zhong Z, Chen W, Gao H et al. Fecal microbiota transplantation exerts a protective role in MPTP-induced Parkinson’s disease via the TLR4/PI3K/AKT/NF-κB pathway stimulated by α-synuclein. Neurochem. Res. 46, 3050–3058 (2021).
189.Sun J, Xu J, Ling Y et al. Fecal microbiota transplantation alleviated Alzheimer’s disease-like pathogenesis in APP/PS1 transgenic mice. Transl. Psychiatry 9(1), 189 (2019).
