فیزیولوژی پزشکی گایتون و هال؛ تحریک ماهیچه اسکلتی

دعای مطالعه [ نمایش ]
بِسْمِ الله الرَّحْمنِ الرَّحیمِ
اَللّهُمَّ اَخْرِجْنى مِنْ ظُلُماتِ الْوَهْمِ
خدايا مرا بيرون آور از تاريكىهاى وهم،
وَ اَكْرِمْنى بِنُورِ الْفَهْمِ
و به نور فهم گرامى ام بدار،
اَللّهُمَّ افْتَحْ عَلَيْنا اَبْوابَ رَحْمَتِكَ
خدايا درهاى رحمتت را به روى ما بگشا،
وَانْشُرْ عَلَيْنا خَزائِنَ عُلُومِكَ بِرَحْمَتِكَ يا اَرْحَمَ الرّاحِمينَ
و خزانههاى علومت را بر ما باز كن به امید رحمتت اى مهربانترين مهربانان.
کتاب «فیزیولوژی پزشکی گایتون و هال» بهعنوان یکی از جامعترین و معتبرترین منابع در حوزه علوم پزشکی، همچنان مرجع کلیدی برای درک عملکرد پیچیده بدن انسان است. این اثر با تکیه بر تازهترین پژوهشها و توضیحات دقیق از سازوکارهای فیزیولوژیک، پلی میان علوم پایه پزشکی و کاربردهای بالینی ایجاد میکند و نقشی بیبدیل در آموزش، پژوهش و ارتقای دانش سلامت ایفا مینماید.
ترجمه دقیق و علمی این شاهکار توسط برند علمی آیندهنگاران مغز به مدیریت داریوش طاهری، دسترسی فارسیزبانان به مرزهای نوین دانش فیزیولوژی را ممکن ساخته و رسالتی علمی برای ارتقای آموزش پزشکی، فهم عمیقتر سازوکارهای بدن و توسعه روشهای نوین در حوزه سلامت فراهم آورده است.
» کتاب فیزیولوژی پزشکی گایتون و هال
» » فصل ۷: تحریک عضله اسکلتی: انتقال عصبی عضلانی و جفت تحریک-انقباض
در حال ویرایش
» Guyton and Hall Textbook of Medical Physiology
»» CHAPTER 7: Excitation of Skeletal Muscle: Neuromuscular Transmission and Excitation-Contraction Coupling

NEUROMUSCULAR JUNCTION AND TRANSMISSION OF IMPULSES FROM NERVE ENDINGS TO SKELETAL MUSCLE FIBERS
Skeletal muscle fibers are innervated by large myelinated nerve fibers that originate from large motoneurons in the anterior horns of the spinal cord. As discussed in Chapter 6, each nerve fiber, after entering the muscle belly, normally branches and stimulates from three to several hundred skeletal muscle fibers. Each nerve ending makes a junction, called the neuromuscular junction, with the muscle fiber near its midpoint. The action potential initi- ated in the muscle fiber by the nerve signal travels in both directions toward the muscle fiber ends. With the exception of about 2% of the muscle fibers, there is only one such junction per muscle fiber.
اتصال عصبی عضلانی و انتقال تکانهها از انتهای عصب به فیبرهای عضلانی اسکلتی
فیبرهای عضلانی اسکلتی توسط رشتههای عصبی میلین دار بزرگی که از نورونهای حرکتی بزرگ در شاخهای قدامینخاع منشا میگیرند، عصب دهی میشوند. همانطور که در فصل 6 بحث شد، هر فیبر عصبی پس از ورود به شکم ماهیچه، به طور معمول از سه تا چند صد فیبر عضلانی اسکلتی منشعب میشود و تحریک میشود. هر انتهای عصبی یک اتصال به نام اتصال عصبی عضلانی را با فیبر عضلانی نزدیک به نقطه میانی خود ایجاد میکند. پتانسیل عمل آغاز شده در فیبر عضلانی توسط سیگنال عصبی در هر دو جهت به سمت انتهای فیبر عضلانی حرکت میکند. به استثنای حدود 2 درصد از فیبرهای عضلانی، تنها یک چنین اتصال در هر فیبر عضلانی وجود دارد.
PHYSIOLOGIC ANATOMY OF THE NEUROMUSCULAR JUNCTION-THE MOTOR END PLATE
Figure 7-1A and B shows the neuromuscular junction from a large myelinated nerve fiber to a skeletal muscle fiber. The nerve fiber forms a complex of branching nerve terminals that invaginate into the surface of the muscle fiber but lie outside the muscle fiber plasma membrane. The entire structure is called the motor end plate. It is covered by one or more Schwann cells that insulate it from the surrounding fluids.
آناتومی فیزیولوژیکی اتصال عصبی-عضلانی- صفحه انتهایی موتور
شکل 7-1A و B اتصال عصبی عضلانی را از یک فیبر عصبی میلین دار بزرگ به یک فیبر عضلانی اسکلتی نشان میدهد. فیبر عصبی مجموعه ای از پایانههای عصبی منشعب را تشکیل میدهد که به سطح فیبر عضلانی نفوذ میکنند اما خارج از غشای پلاسمایی فیبر عضلانی قرار دارند. کل ساختار صفحه انتهایی موتور نامیده میشود. توسط یک یا چند سلول شوان پوشیده شده است که آن را از مایعات اطراف عایق میکند.
Figure 7-1C shows the junction between a single axon terminal and the muscle fiber membrane. The invaginated membrane is called the synaptic gutter or synaptic trough, and the space between the terminal and the fiber membrane is called the synaptic space or synaptic cleft, which is 20 to 30 nanometers wide. At the bottom of the gut- ter are numerous smaller folds of the muscle membrane called subneural clefts, which greatly increase the surface area at which the synaptic transmitter can act.
شکل 7-1C اتصال بین یک پایانه آکسون منفرد و غشای فیبر عضلانی را نشان میدهد. غشای فرورفته ناودان سیناپسی یا ناودان سیناپسی نامیده میشود و فضای بین پایانه و غشای فیبری را فضای سیناپسی یا شکاف سیناپسی مینامند که 20 تا 30 نانومتر عرض دارد. در پایین ناودان، چینهای کوچکتر متعددی از غشای عضلانی به نام شکافهای زیر عصبی وجود دارد که سطحی را که فرستنده سیناپسی میتواند در آن عمل کند، بسیار افزایش میدهد.
In the axon terminal are many mitochondria that supply adenosine triphosphate (ATP), the energy source used for synthesis of a transmitter, acetylcholine, which excites the muscle fiber membrane. Acetylcholine is synthesized in the cytoplasm of the terminal but is absorbed rap- idly into many small synaptic vesicles, about 300,000 of which are normally in the terminals of a single end plate. In the synaptic space are large quantities of the enzyme acetylcholinesterase, which destroys acetylcholine a few milliseconds after it has been released from the synaptic vesicles.
در پایانه آکسون، میتوکندریهای زیادی وجود دارند که آدنوزین تری فسفات (ATP) را تامین میکنند، منبع انرژی که برای سنتز یک فرستنده، استیل کولین، که غشای فیبر عضلانی را تحریک میکند، استفاده میشود. استیل کولین در سیتوپلاسم انتهایی سنتز میشود، اما به سرعت در بسیاری از وزیکولهای سیناپسی کوچک جذب میشود که حدود 300000 تای آن معمولاً در انتهای یک صفحه انتهایی قرار دارند. در فضای سیناپسی مقادیر زیادی آنزیم استیل کولین استراز وجود دارد که استیل کولین را چند میلی ثانیه پس از آزاد شدن از وزیکولهای سیناپسی از بین میبرد.

Figure 7-1.
Different views of the motor end plate. A, Longitudinal section through the end plate. B, Surface view of the end plate. C, Electron micrographic appearance of the contact point between a single axon terminal and the muscle fiber membrane.
شکل 7-1.
نماهای مختلف از صفحه انتهایی موتور A، مقطع طولی از طریق صفحه انتهایی. ب، نمای سطحی صفحه انتهایی. ج، ظاهر میکروگرافیک الکترونی نقطه تماس بین یک پایانه آکسون و غشای فیبر عضلانی.
SECRETION OF ACETYLCHOLINE BY THE NERVE TERMINALS
When a nerve impulse reaches the neuromuscular junc- tion, about 125 vesicles of acetylcholine are released from the terminals into the synaptic space. Some of the details of this mechanism can be seen in Figure 7-2, which shows an expanded view of a synaptic space with the neu- ral membrane above and the muscle membrane and its subneural clefts below.
ترشح استیل کولین توسط پایانههای عصبی
هنگامیکه یک تکانه عصبی به محل اتصال عصبی عضلانی میرسد، حدود 125 وزیکول استیل کولین از پایانهها به فضای سیناپسی آزاد میشود. برخی از جزئیات این مکانیسم را میتوان در شکل 7-2 مشاهده کرد که نمای گسترده ای از فضای سیناپسی را با غشای عصبی در بالا و غشای عضلانی و شکافهای زیر عصبی آن در زیر نشان میدهد.
On the inside surface of the neural membrane are lin- ear dense bars, shown in cross section in Figure 7-2. To each side of each dense bar are protein particles that pen- etrate the neural membrane; these are voltage-gated cal- cium channels. When an action potential spreads over the terminal, these channels open and allow calcium ions to diffuse from the synaptic space to the interior of the nerve terminal. The calcium ions, in turn, are believed to acti- vate Ca2+-calmodulin-dependent protein kinase, which, in turn, phosphorylates synapsin proteins that anchor the acetylcholine vesicles to the cytoskeleton of the pre- synaptic terminal. This process frees the acetylcholine vesicles from the cytoskeleton and allows them to move to the active zone of the presynaptic neural membrane adjacent to the dense bars. The vesicles then dock at the release sites, fuse with the neural membrane, and empty their acetylcholine into the synaptic space by the process of exocytosis.
در سطح داخلی غشای عصبی میلههای متراکم خطی وجود دارد که در شکل 7-2 در مقطع نشان داده شده است. در هر طرف هر نوار متراکم، ذرات پروتئینی وجود دارد که به غشای عصبی نفوذ میکنند. اینها کانالهای کلسیمیبا ولتاژ هستند. هنگامیکه یک پتانسیل عمل روی پایانه پخش میشود، این کانالها باز میشوند و به یونهای کلسیم اجازه میدهند از فضای سیناپسی به داخل انتهای عصبی منتشر شوند. اعتقاد بر این است که یونهای کلسیم به نوبه خود پروتئین کیناز وابسته به Ca2+-calmodulin را فعال میکنند که به نوبه خود پروتئینهای سیناپسین را فسفریله میکند که وزیکولهای استیل کولین را به اسکلت سلولی پایانه پیش سیناپسی متصل میکند. این فرآیند وزیکولهای استیل کولین را از اسکلت سلولی آزاد میکند و به آنها اجازه میدهد تا به منطقه فعال غشای عصبی پیش سیناپسی در مجاورت میلههای متراکم حرکت کنند. سپس وزیکولها در محلهای رهاسازی قرار میگیرند، با غشای عصبی ترکیب میشوند و استیل کولین خود را با فرآیند اگزوسیتوز وارد فضای سیناپسی میکنند.
Although some of the aforementioned details are spec- ulative, it is known that the effective stimulus for causing acetylcholine release from the vesicles is entry of calcium ions and that acetylcholine from the vesicles is then emp- tied through the neural membrane adjacent to the dense bars.
اگرچه برخی از جزئیات ذکر شده حدس و گمان هستند، اما مشخص شده است که محرک موثر برای آزادسازی استیل کولین از وزیکولها، ورود یونهای کلسیم است و سپس استیل کولین از وزیکولها از طریق غشای عصبی مجاور میلههای متراکم تخلیه میشود.

Figure 7-2.
Release of acetylcholine from synaptic vesicles at the neural membrane of the neuromuscular junction. Note the proximity of the release sites in the neural membrane to the acetylcholine recep- tors in the muscle membrane at the mouths of the subneural clefts.
شکل 7-2.
آزادسازی استیل کولین از وزیکولهای سیناپسی در غشای عصبی اتصال عصبی عضلانی. به نزدیکی محلهای رهاسازی در غشای عصبی به گیرندههای استیل کولین در غشای عضلانی در دهانه شکافهای زیر عصبی توجه کنید.
Acetylcholine Opens Ion Channels on Postsynaptic Membranes. Figure 7-2 also shows many small acetyl- choline receptors and voltage-gated sodium channels in the muscle fiber membrane. The acetylcholine-gated ion channels are located almost entirely near the mouths of the subneural clefts lying immediately below the dense bar areas, where the acetylcholine is emptied into the syn- aptic space. The voltage-gated sodium channels also line the subneural clefts.
استیل کولین کانالهای یونی را روی غشاهای پس سیناپسی باز میکند. شکل 7-2 همچنین بسیاری از گیرندههای کوچک استیل کولین و کانالهای سدیم دارای ولتاژ در غشای فیبر عضلانی را نشان میدهد. کانالهای یونی دردار با استیل کولین تقریباً به طور کامل در نزدیکی دهان شکافهای زیر عصبی قرار دارند که بلافاصله در زیر نواحی متراکم میله قرار دارند، جایی که استیل کولین به فضای سیناپسی تخلیه میشود. کانالهای سدیم دارای ولتاژ نیز شکافهای زیر عصبی را میپوشانند.
Each acetylcholine receptor is a protein complex that has a total molecular weight of approximately 275,000. The fetal acetylcholine receptor complex is composed of five subunit proteins, two alpha proteins and one each of beta, delta, and gamma proteins. In the adult, an epsilon protein substitutes for the gamma protein in this recep- tor complex. These protein molecules penetrate all the way through the membrane, lying side by side in a circle to form a tubular channel, illustrated in Figure 7-3. The channel remains constricted, as shown in part A of the figure, until two acetylcholine molecules attach respec- tively to the two alpha subunit proteins. This attachment causes a conformational change that opens the channel, as shown in part B of the figure.
هر گیرنده استیل کولین یک مجموعه پروتئینی است که وزن مولکولی کلی آن تقریباً 275000 است. کمپلکس گیرنده استیل کولین جنین از پنج زیرواحد پروتئین، دو پروتئین آلفا و هر کدام از پروتئینهای بتا، دلتا و گاما تشکیل شده است. در بزرگسالان، یک پروتئین اپسیلون جایگزین پروتئین گاما در این مجتمع گیرنده میشود. این مولکولهای پروتئینی تا آخر غشا نفوذ میکنند و در کنار هم به صورت دایرهای دراز میکشند تا یک کانال لولهای شکل دهند که در شکل 7-3 نشان داده شده است. کانال منقبض میماند، همانطور که در قسمت A شکل نشان داده شده است، تا زمانی که دو مولکول استیل کولین به ترتیب به دو پروتئین زیر واحد آلفا متصل شوند. این پیوست باعث تغییر ساختاری میشود که کانال را باز میکند، همانطور که در قسمت B شکل نشان داده شده است.
The acetylcholine-gated channel has a diameter of about 0.65 nanometer, which is large enough to allow the important positive ions-sodium (Na+), potassium (K+), and calcium (Ca2+)-to move easily through the opening. Patch clamp studies have shown that one of these chan- nels, when opened by acetylcholine, can transmit 15,000 to 30,000 sodium ions in 1 millisecond. Conversely, nega- tive ions, such as chloride ions, do not pass through because of strong negative charges in the mouth of the channel that repel these negative ions.
کانال دریچه ای استیل کولین دارای قطری در حدود 0.65 نانومتر است که به اندازه کافی بزرگ است که به یونهای مثبت مهم سدیم (Na+)، پتاسیم (K+) و کلسیم (Ca2+) اجازه میدهد تا به راحتی از طریق دهانه حرکت کنند. مطالعات پچ گیره نشان داده است که یکی از این کانالها، زمانی که توسط استیل کولین باز میشود، میتواند 15000 تا 30000 یون سدیم را در 1 میلی ثانیه منتقل کند. برعکس، یونهای منفی، مانند یونهای کلرید، به دلیل بارهای منفی قوی در دهانه کانال که این یونهای منفی را دفع میکنند، عبور نمیکنند.
In practice, far more sodium ions flow through the acetylcholine-gated channels than any other ions for two reasons. First, there are only two positive ions present in large concentrations-sodium ions in the extracellular fluid and potassium ions in the intracel- lular fluid. Second, the negative potential on the inside of the muscle membrane, -80 to -90 millivolts, pulls the positively charged sodium ions to the inside of the fiber while simultaneously preventing efflux of the pos- itively charged potassium ions when they attempt to pass outward.
در عمل، به دو دلیل، یونهای سدیم به مراتب بیشتر از هر یون دیگری در کانالهای دریچه ای استیل کولین جریان مییابد. اول، تنها دو یون مثبت در غلظتهای زیاد وجود دارد: یون سدیم در مایع خارج سلولی و یون پتاسیم در مایع داخل سلولی. دوم، پتانسیل منفی در داخل غشای عضلانی، 80- تا 90- میلی ولت، یونهای سدیم با بار مثبت را به داخل فیبر میکشد و همزمان از خروج یونهای پتاسیم با بار مثبت در هنگام عبور از بیرون جلوگیری میکند.
As shown in Figure 7-3B, the principal effect of open- ing the acetylcholine-gated channels is to allow sodium ions to flow to the inside of the fiber, carrying positive charges with them. This action creates a local positive potential change inside the muscle fiber membrane, called the end plate potential. This end plate potential normally causes sufficient depolarization to open neighboring voltage-gated sodium channels, allowing even greater sodium ion inflow and initiating an action potential that spreads along the muscle membrane and causes muscle contraction.
همانطور که در شکل 7-3B نشان داده شده است، اثر اصلی باز کردن کانالهای دریچه ای استیل کولین این است که به یونهای سدیم اجازه میدهد تا به داخل فیبر جریان پیدا کنند و بارهای مثبت را با خود حمل کنند. این عمل یک تغییر پتانسیل مثبت موضعی در داخل غشای فیبر عضلانی ایجاد میکند که به آن پتانسیل صفحه انتهایی میگویند. این پتانسیل صفحه انتهایی معمولاً باعث دپلاریزاسیون کافی برای بازکردن کانالهای سدیم دارای ولتاژ همسایه میشود و اجازه میدهد حتی بیشتر وارد یون سدیم شود و پتانسیل عملی را آغاز میکند که در امتداد غشای عضلانی پخش میشود و باعث انقباض عضلانی میشود.
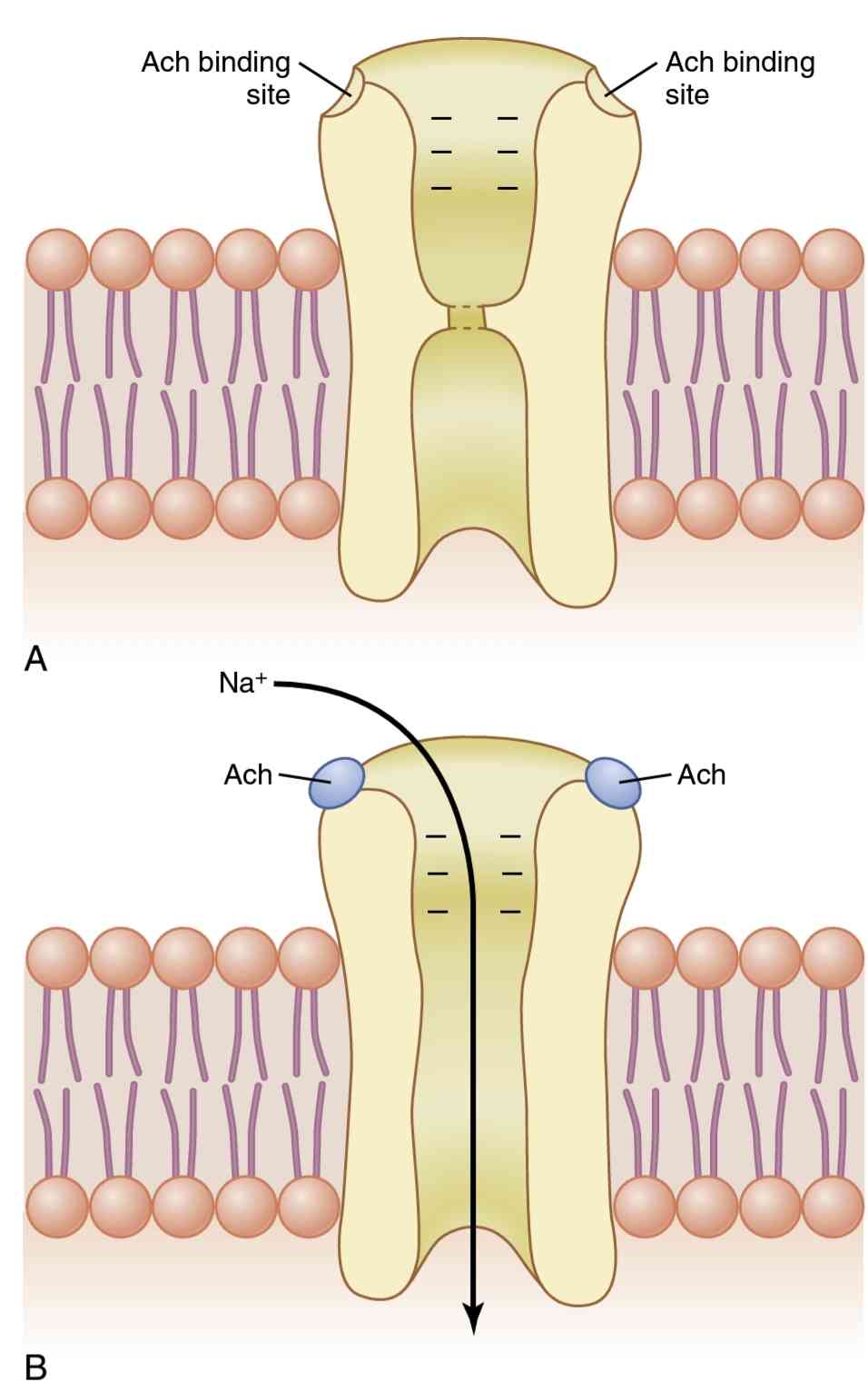
Figure 7-3.
Acetylcholine-gated channel. A, Closed state. B, After acetylcholine (Ach) has become attached and a conformational change has opened the channel, allowing sodium ions to enter the muscle fiber and excite contraction. Note the negative charges at the channel mouth that prevent passage of negative ions such as chloride ions.
شکل 7-3.
کانال دردار با استیل کولین الف، حالت بسته B، پس از اینکه استیل کولین (Ach) متصل شد و یک تغییر ساختاری کانال را باز کرد، اجازه میدهد یونهای سدیم وارد فیبر عضلانی شده و انقباض را تحریک کنند. به بارهای منفی در دهانه کانال توجه کنید که از عبور یونهای منفی مانند یونهای کلرید جلوگیری میکند.
Destruction of the Released Acetylcholine by Ace- tylcholinesterase. The acetylcholine, once released into the synaptic space, continues to activate acetylcholine re- ceptors as long as the acetylcholine persists in the space. However, it is rapidly destroyed by the enzyme acetylcho- linesterase, which is attached mainly to the spongy layer of fine connective tissue that fills the synaptic space be- tween the presynaptic nerve terminal and the postsynap- tic muscle membrane. A small amount of acetylcholine diffuses out of the synaptic space and is then no longer available to act on the muscle fiber membrane.
تخریب استیل کولین آزاد شده توسط استیل کولین استراز. استیل کولین، پس از آزاد شدن در فضای سیناپسی، تا زمانی که استیل کولین در فضا باقی بماند، به فعال کردن گیرندههای استیل کولین ادامه میدهد. با این حال، به سرعت توسط آنزیم استیل کولین استراز، که عمدتاً به لایه اسفنجی بافت همبند ظریفی که فضای سیناپسی بین پایانه عصبی پیش سیناپسی و غشای عضلانی پس سیناپسی را پر میکند، متصل میشود، از بین میرود. مقدار کمیاستیل کولین از فضای سیناپسی پخش میشود و دیگر برای اثرگذاری بر روی غشای فیبر عضلانی در دسترس نیست.
The short time that the acetylcholine remains in the synaptic space-a few milliseconds at most-normally is sufficient to excite the muscle fiber. Then the rapid removal of the acetylcholine prevents continued muscle re-excitation after the muscle fiber has recovered from its initial action potential.
مدت زمان کوتاهی که استیل کولین در فضای سیناپسی باقی میماند – در حد اکثر چند میلی ثانیه – در حالت عادی برای تحریک فیبر عضلانی کافی است. سپس حذف سریع استیل کولین از ادامه تحریک مجدد ماهیچه پس از بازیابی فیبر عضلانی از پتانسیل عمل اولیه خود جلوگیری میکند.
End Plate Potential and Excitation of the Skeletal Muscle Fiber. The sudden insurgence of sodium ions into the muscle fiber when the acetylcholine-gated chan- nels open causes the electrical potential inside the fiber at the local area of the end plate to increase in the positive direction as much as 50 to 75 millivolts, creating a local potential called the end plate potential. Recall from Chap- ter 5 that a sudden increase in nerve membrane potential of more than 20 to 30 millivolts is normally sufficient to initiate more and more sodium channel opening, thus ini- tiating an action potential at the muscle fiber membrane.
پتانسیل صفحه انتهایی و تحریک فیبر عضلانی اسکلتی. هجوم ناگهانی یونهای سدیم به فیبر عضلانی هنگام باز شدن کانالهای دریچه ای استیل کولین باعث میشود که پتانسیل الکتریکی داخل فیبر در ناحیه محلی صفحه انتهایی در جهت مثبت به میزان 50 تا 75 میلی ولت افزایش یابد و پتانسیل محلی به نام پتانسیل صفحه انتهایی ایجاد شود. از فصل 5 به یاد بیاورید که افزایش ناگهانی پتانسیل غشای عصبی بیش از 20 تا 30 میلی ولت معمولاً برای شروع بیشتر و بیشتر باز شدن کانال سدیم کافی است، بنابراین یک پتانسیل عمل در غشای فیبر عضلانی آغاز میشود.
Figure 7-4 illustrates an end plate potential initiat- ing the action potential. This figure shows three separate end plate potentials. End plate potentials A and C are too weak to elicit an action potential, but they do pro- duce weak local end plate voltage changes, as recorded in the figure. By contrast, end plate potential B is much stronger and causes enough sodium channels to open so that the self-regenerative effect of more and more sodium ions flowing to the interior of the fiber initiates an action potential. The weakness of the end plate potential at point A was caused by poisoning of the muscle fiber with curare, a drug that blocks the gating action of ace- tylcholine on the acetylcholine channels by competing for the acetylcholine receptor sites. The weakness of the end plate potential at point C resulted from the effect of botu- linum toxin, a bacterial poison that decreases the quantity of acetylcholine release by the nerve terminals.
شکل 7-4 یک پتانسیل صفحه انتهایی را نشان میدهد که پتانسیل عمل را آغاز میکند. این شکل سه پتانسیل صفحه انتهایی مجزا را نشان میدهد. پتانسیلهای صفحه انتهایی A و C برای ایجاد پتانسیل عمل بسیار ضعیف هستند، اما همانطور که در شکل ثبت شده است، تغییرات محلی ضعیف ولتاژ صفحه انتهایی ایجاد میکنند. در مقابل، پتانسیل صفحه انتهایی B بسیار قویتر است و باعث میشود کانالهای سدیم به اندازه کافی باز شوند، به طوری که اثر خودبازسازی یونهای سدیم بیشتر و بیشتر که به داخل فیبر جریان مییابند، پتانسیل عمل را آغاز میکند. ضعف پتانسیل صفحه انتهایی در نقطه A ناشی از مسمومیت فیبر عضلانی با کورار است، دارویی که با رقابت برای مکانهای گیرنده استیل کولین، مانع از عملکرد دروازهای استیل کولین در کانالهای استیل کولین میشود. ضعف پتانسیل صفحه انتهایی در نقطه C ناشی از اثر سم بوتولینوم، یک سم باکتریایی است که مقدار آزاد شدن استیل کولین توسط پایانههای عصبی را کاهش میدهد.

Figure 7-4.
End plate potentials (in millivolts). A, Weakened end plate potential recorded in a curarized muscle that is too weak to elicit an action potential. B, Normal end plate potential eliciting a muscle action potential. C, Weakened end plate potential caused by botulinum toxin that decreases end plate release of acetylcholine, again too weak to elicit a muscle action potential.
شکل 7-4.
پتانسیل صفحه انتهایی (به میلی ولت). الف، پتانسیل صفحه انتهایی ضعیف شده در یک عضله کوراریزه شده ثبت شده است که برای برانگیختن پتانسیل عمل بسیار ضعیف است. B، پتانسیل صفحه انتهایی طبیعی که پتانسیل عمل عضلانی را برمیانگیزد. C، ضعیف شدن پتانسیل صفحه انتهایی ناشی از سم بوتولینوم که آزادسازی استیل کولین در صفحه انتهایی را کاهش میدهد، دوباره برای ایجاد پتانسیل عمل عضلانی ضعیف تر از آن است.
Safety Factor for Transmission at the Neuromuscu- lar Junction-Fatigue of the Junction. Ordinarily, each impulse that arrives at the neuromuscular junction causes about three times as much end plate potential as that required to stimulate the muscle fiber. Therefore, the normal neuromuscular junction is said to have a high safety factor. However, stimulation of the nerve fiber at rates greater than 100 times per second for several min- utes may diminish the number of acetylcholine vesicles so much that impulses fail to pass into the muscle fiber. This situation is called fatigue of the neuromuscular junction, and it is the same effect that causes fatigue of synapses in the central nervous system when the synapses are overex- cited. Under normal functioning conditions, measurable fatigue of the neuromuscular junction occurs rarely and, even then, only at the most exhausting levels of muscle activity.
فاکتور ایمنی برای انتقال در محل اتصال عصبی عضلانی-خستگی محل اتصال. به طور معمول، هر تکانه ای که به محل اتصال عصبی عضلانی میرسد، حدود سه برابر بیشتر از پتانسیل صفحه انتهایی مورد نیاز برای تحریک فیبر عضلانی ایجاد میکند. بنابراین گفته میشود که اتصال عصبی عضلانی طبیعی دارای ضریب ایمنی بالایی است. با این حال، تحریک فیبر عصبی با سرعت بیش از 100 بار در ثانیه برای چند دقیقه ممکن است تعداد وزیکولهای استیل کولین را به قدری کاهش دهد که تکانهها به فیبر عضلانی منتقل نشوند. این وضعیت خستگی اتصال عصبی عضلانی نامیده میشود و همان اثری است که باعث خستگی سیناپسها در سیستم عصبی مرکزی در هنگام تحریک بیش از حد سیناپسها میشود. در شرایط عملکرد نرمال، خستگی قابل اندازه گیری اتصال عصبی عضلانی به ندرت و حتی در آن زمان تنها در طاقت فرساترین سطوح فعالیت عضلانی رخ میدهد.
Acetylcholine Formation and Release
Acetylcholine formation and release at the neuromuscular junction occur in the following stages:
تشکیل و آزادسازی استیل کولین
تشکیل و آزادسازی استیل کولین در محل اتصال عصبی عضلانی در مراحل زیر رخ میدهد:
1. Small vesicles, about 40 nanometers in size, are formed by the Golgi apparatus in the cell body of the motoneu- ron in the spinal cord. These vesicles are then trans- ported by axoplasm that streams through the core of the axon from the central cell body in the spinal cord all the way to the neuromuscular junction at the tips of the peripheral nerve fibers. About 300,000 of these small vesicles collect in the nerve terminals of a single skeletal muscle end plate.
2. Acetylcholine is synthesized in the cytosol of the nerve fiber terminal but is immediately transported through the membranes of the vesicles to their interior, where it is stored in highly concentrated form-about 10,000 molecules of acetylcholine in each vesicle.
3. When an action potential arrives at the nerve terminal, it opens many calcium channels in the membrane of the nerve terminal because this terminal has an abundance of voltage-gated calcium channels. As a result, the cal- cium ion concentration inside the terminal membrane increases about 100-fold, which in turn increases the rate of fusion of the acetylcholine vesicles with the ter- minal membrane about 10,000-fold. This fusion makes many of the vesicles rupture, allowing exocytosis of ace- tylcholine into the synaptic space. About 125 vesicles usually rupture with each action potential. Then, after a few milliseconds, the acetylcholine is split by acetylcho- linesterase into acetate ion and choline, and the choline is actively reabsorbed into the neural terminal to be re- used to form new acetylcholine. This sequence of events occurs within a period of 5 to 10 milliseconds.
4. The number of vesicles available in the nerve ending is sufficient to allow transmission of only a few thousand nerve to muscle impulses. Therefore, for continued function of the neuromuscular junction, new vesicles need to be re-formed rapidly. Within a few seconds after each action potential is over, coated pits appear in the terminal nerve membrane, caused by contractile pro- teins in the nerve ending, especially the protein clathrin, which is attached to the membrane in the areas of the original vesicles. Within about 20 seconds, the proteins contract and cause the pits to break away to the interior of the membrane, thus forming new vesicles. Within another few seconds, acetylcholine is transported to the interior of these vesicles, and they are then ready for a new cycle of acetylcholine release.
1. وزیکولهای کوچک با اندازه حدود 40 نانومتر توسط دستگاه گلژی در بدنه سلولی حرکتی در طناب نخاعی تشکیل میشوند. این وزیکولها سپس توسط آکسوپلاسم منتقل میشوند که از طریق هسته آکسون از بدن سلول مرکزی در نخاع تا نقطه اتصال عصبی عضلانی در نوک رشتههای عصبی محیطی جریان مییابد. حدود 300000 از این وزیکولهای کوچک در پایانههای عصبی یک صفحه انتهایی عضله اسکلتی جمع میشوند.
2. استیل کولین در سیتوزول پایانه فیبر عصبی سنتز میشود، اما بلافاصله از طریق غشای وزیکولها به داخل آنها منتقل میشود، جایی که به شکل بسیار غلیظ – حدود 10000 مولکول استیل کولین در هر وزیکول ذخیره میشود.
3. هنگامیکه یک پتانسیل عمل به پایانه عصبی میرسد، بسیاری از کانالهای کلسیمیرا در غشای پایانه عصبی باز میکند زیرا این پایانه دارای کانالهای کلسیمیبا ولتاژ فراوان است. در نتیجه، غلظت یون کلسیم در داخل غشای انتهایی حدود 100 برابر افزایش مییابد که به نوبه خود سرعت همجوشی وزیکولهای استیل کولین با غشای انتهایی را حدود 10000 برابر افزایش میدهد. این همجوشی باعث میشود که بسیاری از وزیکولها پاره شوند و به اگزوسیتوز استیل کولین وارد فضای سیناپسی شود. معمولاً با هر پتانسیل عمل حدود 125 وزیکول پاره میشود. سپس، پس از چند میلی ثانیه، استیل کولین توسط استیل کولین استراز به یون استات و کولین تقسیم میشود و کولین به طور فعال در پایانه عصبی بازجذب میشود تا دوباره برای تشکیل استیل کولین جدید استفاده شود. این توالی رویدادها در بازه زمانی 5 تا 10 میلی ثانیه رخ میدهد.
4. تعداد وزیکولهای موجود در انتهای عصب برای انتقال تنها چند هزار تکانه عصبی به ماهیچه کافی است. بنابراین، برای ادامه عملکرد اتصال عصبی عضلانی، وزیکولهای جدید باید به سرعت دوباره تشکیل شوند. در عرض چند ثانیه پس از اتمام هر پتانسیل عمل، حفرههای پوشیدهشده در غشای عصب انتهایی ظاهر میشوند که ناشی از پروتئینهای انقباضی در انتهای عصب، بهویژه پروتئین کلاترین است که در نواحی وزیکولهای اصلی به غشاء متصل است. در عرض حدود 20 ثانیه، پروتئینها منقبض میشوند و باعث میشوند که حفرهها به داخل غشاء برسند و در نتیجه وزیکولهای جدیدی تشکیل شود. در عرض چند ثانیه دیگر، استیل کولین به داخل این وزیکولها منتقل میشود و آنها برای چرخه جدید آزادسازی استیل کولین آماده میشوند.
Drugs That Enhance or Block Transmission at the Neuromuscular Junction
داروهایی که انتقال را در محل اتصال عصبی عضلانی تقویت یا مسدود میکنند
Drugs That Stimulate the Muscle Fiber by Acetylcholine- Like Action. Several compounds, including methacholine, carbachol, and nicotine, have nearly the same effect on the muscle fiber as acetylcholine. The main differences be- tween these drugs and acetylcholine are that the drugs are not destroyed by cholinesterase or are destroyed so slowly that their action often persists for many minutes to several hours. The drugs work by causing localized areas of depo- larization of the muscle fiber membrane at the motor end plate where the acetylcholine receptors are located. Then, every time the muscle fiber recovers from a previous con- traction, these depolarized areas, by virtue of leaking ions, initiate a new action potential, thereby causing a state of muscle spasm.
داروهایی که فیبر عضلانی را با استیل کولین تحریک میکنند. چندین ترکیب از جمله متاکولین، کارباکول و نیکوتین تقریباً همان تأثیر استیل کولین را بر روی فیبر عضلانی دارند. تفاوت اصلی بین این داروها و استیل کولین در این است که داروها توسط کولین استراز از بین نمیروند یا آنقدر آهسته از بین میروند که اثر آنها اغلب برای چندین دقیقه تا چند ساعت ادامه دارد. داروها با ایجاد نواحی موضعی دپولاریزاسیون غشای فیبر عضلانی در صفحه انتهایی حرکتی که گیرندههای استیل کولین در آن قرار دارند، عمل میکنند. سپس، هر بار که فیبر عضلانی از یک انقباض قبلی بهبود مییابد، این نواحی دپلاریزه شده، به دلیل نشت یونها، پتانسیل عمل جدیدی را آغاز میکنند و در نتیجه باعث ایجاد حالت اسپاسم عضلانی میشوند.
Drugs That Stimulate the Neuromuscular Junction by Inactivating Acetylcholinesterase. Three particularly well-known drugs-neostigmine, physostigmine, and diiso- propyl fluorophosphate-inactivate acetylcholinesterase in the synapses so that it no longer hydrolyzes acetylcholine. Therefore, with each successive nerve impulse, additional acetylcholine accumulates and stimulates the muscle fiber repetitively. This activity causes muscle spasm when even a few nerve impulses reach the muscle. Unfortunately, it can also cause death as a result of laryngeal spasm, which smothers a person.
داروهایی که اتصال عصبی عضلانی را با غیرفعال کردن استیل کولین استراز تحریک میکنند. سه داروی شناخته شده به ویژه نئوستیگمین، فیزوستیگمین و دی ایزوپروپیل فلوروفسفات استیل کولین استراز را غیرفعال میکنند به طوری که دیگر استیل کولین را هیدرولیز نمیکند. بنابراین، با هر تکانه عصبی متوالی، استیل کولین اضافی انباشته شده و فیبر عضلانی را به طور مکرر تحریک میکند. این فعالیت زمانی که حتی چند تکانه عصبی به عضله برسد باعث اسپاسم عضلانی میشود. متأسفانه، همچنین میتواند در نتیجه اسپاسم حنجره که فرد را خفه میکند، منجر به مرگ شود.
Neostigmine and physostigmine combine with acetyl- cholinesterase to inactivate the acetylcholinesterase for up to several hours, after which these drugs are displaced from the acetylcholinesterase so that the esterase once again be- comes active. Conversely, diisopropyl fluorophosphate, which is a powerful nerve gas poison, inactivates acetylcho- linesterase for weeks, which makes this poison particularly lethal.
نئوستیگمین و فیزوستیگمین با استیل کولین استراز ترکیب میشوند تا استیل کولین استراز را تا چند ساعت غیرفعال کنند و پس از آن این داروها از استیل کولین استراز جابجا میشوند تا استراز دوباره فعال شود. برعکس، دی ایزوپروپیل فلوروفسفات، که یک سم قوی گاز عصبی است، استیل کولین استراز را برای هفتهها غیرفعال میکند که این سم را به ویژه کشنده میکند.
Drugs That Block Transmission at the Neuromuscular Junction. A group of drugs known as curariform drugs can prevent the passage of impulses from the nerve ending into the muscle. For example. D-tubocurarine blocks the action of acetylcholine on the muscle fiber acetylcholine receptors, thus preventing sufficient increase in permeability of the muscle membrane channels to initiate an action potential.
داروهایی که انتقال را در محل اتصال عصبی عضلانی مسدود میکنند. گروهی از داروها که به عنوان داروهای کوراریفرم شناخته میشوند، میتوانند از عبور تکانهها از انتهای عصب به عضله جلوگیری کنند. به عنوان مثال. D-tubocurarine از عمل استیل کولین بر روی گیرندههای استیل کولین فیبر عضلانی جلوگیری میکند، بنابراین از افزایش کافی در نفوذپذیری کانالهای غشای عضلانی برای شروع پتانسیل عمل جلوگیری میکند.
Myasthenia Gravis Causes Muscle Weakness
Myasthenia gravis, which occurs in about 1 in every 20,000 persons, causes muscle weakness because of the inability of the neuromuscular junctions to transmit enough signals from the nerve fibers to the muscle fibers. Antibodies that attack the acetylcholine receptors have been demonstrat- ed in the blood of most patients with myasthenia gravis. Therefore, myasthenia gravis is believed to be an autoim- mune disease in which the patients have developed anti- bodies that block or destroy their own acetylcholine recep- tors at the postsynaptic neuromuscular junction.
میاستنی گراویس باعث ضعف عضلانی میشود
میاستنی گراویس که در حدود 1 نفر از هر 20000 نفر رخ میدهد، به دلیل ناتوانی اتصالات عصبی عضلانی در انتقال سیگنالهای کافی از رشتههای عصبی به فیبرهای عضلانی، باعث ضعف عضلانی میشود. آنتی بادیهایی که به گیرندههای استیل کولین حمله میکنند در خون اکثر بیماران مبتلا به میاستنی گراویس نشان داده شده است. بنابراین اعتقاد بر این است که میاستنی گراویس یک بیماری خودایمنی است که در آن بیماران آنتیبادیهایی ایجاد کردهاند که گیرندههای استیل کولین خود را در محل اتصال عصبی عضلانی پس سیناپسی مسدود یا تخریب میکنند.
Regardless of the cause, the end plate potentials that occur in the muscle fibers are mostly too weak to initiate opening of the voltage-gated sodium channels, and muscle fiber depolarization does not occur. If the disease is intense enough, the patient may die of respiratory failure as a result of severe weakness of the respiratory muscles. The disease can usually be ameliorated for several hours by adminis- tering neostigmine or some other anticholinesterase drug, which allows larger than normal amounts of acetylcholine to accumulate in the synaptic space. Within minutes, some of those affected can begin to function almost normally un- til a new dose of neostigmine is required a few hours later.
صرف نظر از علت، پتانسیل صفحه انتهایی که در فیبرهای عضلانی رخ میدهد، اغلب برای شروع باز شدن کانالهای سدیم دارای ولتاژ ضعیف هستند و دپلاریزاسیون فیبر عضلانی رخ نمیدهد. اگر بیماری به اندازه کافی شدید باشد، ممکن است بیمار بر اثر نارسایی تنفسی در نتیجه ضعف شدید عضلات تنفسی بمیرد. این بیماری معمولاً میتواند برای چند ساعت با تجویز نئوستیگمین یا برخی داروهای آنتی کولین استراز دیگر بهبود یابد، که اجازه میدهد مقادیر بیش از حد طبیعی استیل کولین در فضای سیناپسی تجمع یابد. در عرض چند دقیقه، برخی از افراد مبتلا میتوانند تقریباً به طور طبیعی شروع به عملکرد کنند تا اینکه چند ساعت بعد دوز جدید نئوستیگمین مورد نیاز است.
MUSCLE ACTION POTENTIAL
Almost everything discussed in Chapter 5 regarding the initiation and conduction of action potentials in nerve fibers applies equally to skeletal muscle fibers, except for quantitative differences. Some of the quantitative aspects of muscle potentials are as follows:
پتانسیل عمل عضلانی
تقریباً همه چیزهایی که در فصل 5 در مورد شروع و هدایت پتانسیلهای عمل در رشتههای عصبی مورد بحث قرار گرفت، به جز تفاوتهای کمی، در مورد فیبرهای عضلانی اسکلتی نیز صدق میکند. برخی از جنبههای کمیپتانسیل عضلانی به شرح زیر است:
1. The resting membrane potential is about -80 to -90 millivolts in skeletal fibers, about 10 to 20 millivolts more negative than in neurons.
2. The duration of the action potential is 1 to 5 mil- liseconds in skeletal muscle, about five times as long as in large myelinated nerves.
3. The velocity of conduction is 3 to 5 m/sec, about 1/13 the velocity of conduction in the large myeli- nated nerve fibers that excite skeletal muscle.
1. پتانسیل غشای در حال استراحت در الیاف اسکلتی حدود 80- تا 90- میلی ولت است، حدود 10 تا 20 میلی ولت منفی تر از نورونها.
2. مدت زمان پتانسیل عمل در عضله اسکلتی 1 تا 5 میلی ثانیه است، تقریباً پنج برابر طولانی تر از اعصاب میلین دار بزرگ.
3. سرعت رسانش 3 تا 5 متر بر ثانیه است، یعنی حدود 1/13 سرعت رسانش در رشتههای عصبی بزرگ میلین شده که ماهیچههای اسکلتی را تحریک میکنند.
Action Potentials Spread to the Interior of the Muscle Fiber by Way of Transverse Tubules
The skeletal muscle fiber is so large that action poten- tials spreading along its surface membrane cause almost no current flow deep within the fiber. Maximum muscle contraction, however, requires the current to penetrate deeply into the muscle fiber to the vicinity of the separate myofibrils. This penetration is achieved by transmission of action potentials along transverse tubules (T tubules) that penetrate all the way through the muscle fiber, from one side of the fiber to the other, as illustrated in Figure 7-5. The T tubule action poten- tials cause release of calcium ions inside the muscle fiber in the immediate vicinity of the myofibrils, and these calcium ions then cause contraction. The overall process is called excitation-contraction coupling.
پتانسیلهای عمل از طریق لولههای عرضی به داخل فیبر عضلانی گسترش مییابد
فیبر عضلانی اسکلتی آنقدر بزرگ است که پتانسیلهای عملی که در امتداد غشای سطحی آن پخش میشوند باعث میشوند تقریباً هیچ جریانی در عمق فیبر جریان نداشته باشد. با این حال، حداکثر انقباض عضلانی نیاز به نفوذ جریان عمیق به فیبر عضلانی در مجاورت میوفیبریلهای جداگانه دارد. این نفوذ با انتقال پتانسیلهای عمل در امتداد لولههای عرضی (توبولهای T) به دست میآید که در تمام طول فیبر عضلانی، از یک طرف فیبر به سمت دیگر نفوذ میکنند، همانطور که در شکل 7-5 نشان داده شده است. پتانسیل عمل توبول T باعث آزاد شدن یونهای کلسیم در داخل فیبر عضلانی در مجاورت میوفیبریلها میشود و این یونهای کلسیم باعث انقباض میشوند. فرآیند کلی جفت تحریک – انقباض نامیده میشود.

Figure 7-5.
Transverse (T) tubule-sarcoplasmic reticulum system. Note that the T tubules communicate with the outside of the cell membrane and, deep in the muscle fiber, each T tubule lies adjacent to the ends of longitudinal sarcoplasmic reticulum tubules that surround all sides of the actual myofibrils that contract. This illustration was drawn from frog muscle, which has one T tubule per sarcomere, located at the Z disk. A similar arrangement is found in mammalian heart muscle, but mammalian skeletal muscle has two T tubules per sarcomere, located at the A-l band junctions.
شکل 7-5.
سیستم شبکه لوله سارکوپلاسمیعرضی (T). توجه داشته باشید که لولههای T با خارج از غشای سلولی ارتباط برقرار میکنند و در اعماق فیبر عضلانی، هر لوله T در مجاورت انتهای لولههای شبکه سارکوپلاسمیطولی قرار میگیرد که همه طرفهای میوفیبریلهای واقعی را که منقبض میشوند احاطه کرده اند. این تصویر از ماهیچه قورباغه گرفته شده است که دارای یک لوله T در هر سارکومر است که در دیسک Z قرار دارد. آرایش مشابهی در عضله قلب پستانداران یافت میشود، اما عضله اسکلتی پستانداران دارای دو لوله T در هر سارکومر است که در اتصالات باند A-l قرار دارد.
EXCITATION-CONTRACTION COUPLING
کوپلینگ تحریک-انقباض
Transverse Tubule-Sarcoplasmic Reticulum System
Figure 7-5 shows myofibrils surrounded by the T tubule- sarcoplasmic reticulum system. The T tubules are small and run transverse to the myofibrils. They begin at the cell membrane and penetrate all the way from one side of the muscle fiber to the opposite side. Not shown in the figure is that these tubules branch among themselves and form entire planes of T tubules interlacing among all the separate myofibrils. Also, where the T tubules originate from the cell membrane, they are open to the exterior of the muscle fiber. Therefore, they communicate with the extracellular fluid surrounding the muscle fiber and con- tain extracellular fluid in their lumens. In other words, the T tubules are actually internal extensions of the cell mem- brane. Therefore, when an action potential spreads over a muscle fiber membrane, a potential change also spreads along the T tubules to the deep interior of the muscle fiber. The electrical currents surrounding these T tubules then elicit the muscle contraction.
سیستم شبکه لوله ای عرضی-سارکوپلاسمی
شکل 7-5 میوفیبریلهای احاطه شده توسط لوله T- سیستم شبکه سارکوپلاسمیرا نشان میدهد. لولههای T کوچک هستند و به صورت عرضی به میوفیبریلها کشیده میشوند. آنها از غشای سلولی شروع میشوند و از یک طرف فیبر عضلانی به طرف مقابل نفوذ میکنند. در شکل نشان داده نشده است که این لولهها در بین خود منشعب میشوند و صفحات کاملی از لولههای T را تشکیل میدهند که در بین تمام میوفیبریلهای جداگانه در هم آمیخته شده اند. همچنین، در جایی که لولههای T از غشای سلولی منشا میگیرند، به سمت بیرونی فیبر عضلانی باز میشوند. بنابراین، آنها با مایع خارج سلولی اطراف فیبر عضلانی ارتباط برقرار میکنند و حاوی مایع خارج سلولی در لومن خود هستند. به عبارت دیگر، لولههای T در واقع امتداد داخلی غشای سلولی هستند. بنابراین، هنگامیکه یک پتانسیل عمل روی غشای فیبر عضلانی پخش میشود، یک تغییر پتانسیل نیز در امتداد لولههای T به داخل عمیق فیبر عضلانی گسترش مییابد. سپس جریان الکتریکی اطراف این لولههای T باعث انقباض عضلانی میشود.
Figure 7-5 also shows a sarcoplasmic reticulum, in yellow. This sarcoplasmic reticulum is composed of two major parts: (1) large chambers called terminal cisternae that abut the T tubules; and (2) long longitudinal tubules that surround all surfaces of the contracting myofibrils.
شکل 7-5 همچنین یک شبکه سارکوپلاسمیرا به رنگ زرد نشان میدهد. این شبکه سارکوپلاسمیاز دو بخش اصلی تشکیل شده است: (1) محفظههای بزرگی به نام سیسترناهای انتهایی که به لولههای T متصل میشوند. و (2) لولههای طولی طولانی که تمام سطوح میوفیبریلهای منقبض را احاطه کرده اند.
Release of Calcium lons by the Sarcoplasmic Reticulum
One of the special features of the sarcoplasmic reticulum is that within its vesicular tubules is an excess of calcium ions in high concentration. Many of these ions are released from each vesicle when an action potential occurs in the adjacent T tubule.
آزادسازی لان کلسیم توسط شبکه سارکوپلاسمی
یکی از ویژگیهای خاص شبکه سارکوپلاسمیاین است که در داخل لولههای تاولی آن، یونهای کلسیم اضافی در غلظت بالا وجود دارد. بسیاری از این یونها از هر وزیکول زمانی که پتانسیل عمل در لوله T مجاور رخ میدهد آزاد میشوند.
Figures 7-6 and 7-7 show that the action potential of the T tubule causes current flow into the sarcoplasmic reticular cisternae where they abut the T tubule. As the action potential reaches the T tubule, the voltage change is sensed by dihydropyridine receptors linked to calcium release channels, also called ryanodine receptor channels, in the adjacent sarcoplasmic reticular cisternae (see Fig- ure 7-6). Activation of dihydropyridine receptors triggers the opening of the calcium release channels in the cister- nae, as well as in their attached longitudinal tubules. These channels remain open for a few milliseconds, releasing calcium ions into the sarcoplasm surrounding the myo- fibrils and causing contraction, as discussed in Chapter 6.
شکلهای 7-6 و 7-7 نشان میدهند که پتانسیل عمل لوله T باعث میشود جریان به داخل مخزن شبکه سارکوپلاسمیجایی که به لوله T متصل میشوند، جریان یابد. هنگامیکه پتانسیل عمل به لوله T میرسد، تغییر ولتاژ توسط گیرندههای دی هیدروپیریدینی مرتبط با کانالهای رهاسازی کلسیم، که کانالهای گیرنده رایانودین نیز نامیده میشوند، در مخزن شبکه سارکوپلاسمیمجاور حس میشود (شکل 7-6 را ببینید). فعال شدن گیرندههای دی هیدروپیریدینی باعث باز شدن کانالهای آزادسازی کلسیم در مخزن و همچنین در لولههای طولی متصل به آنها میشود. این کانالها برای چند میلی ثانیه باز میمانند و یونهای کلسیم را در سارکوپلاسم اطراف میوفیبریلها آزاد میکنند و باعث انقباض میشوند، همانطور که در فصل 6 بحث شد.
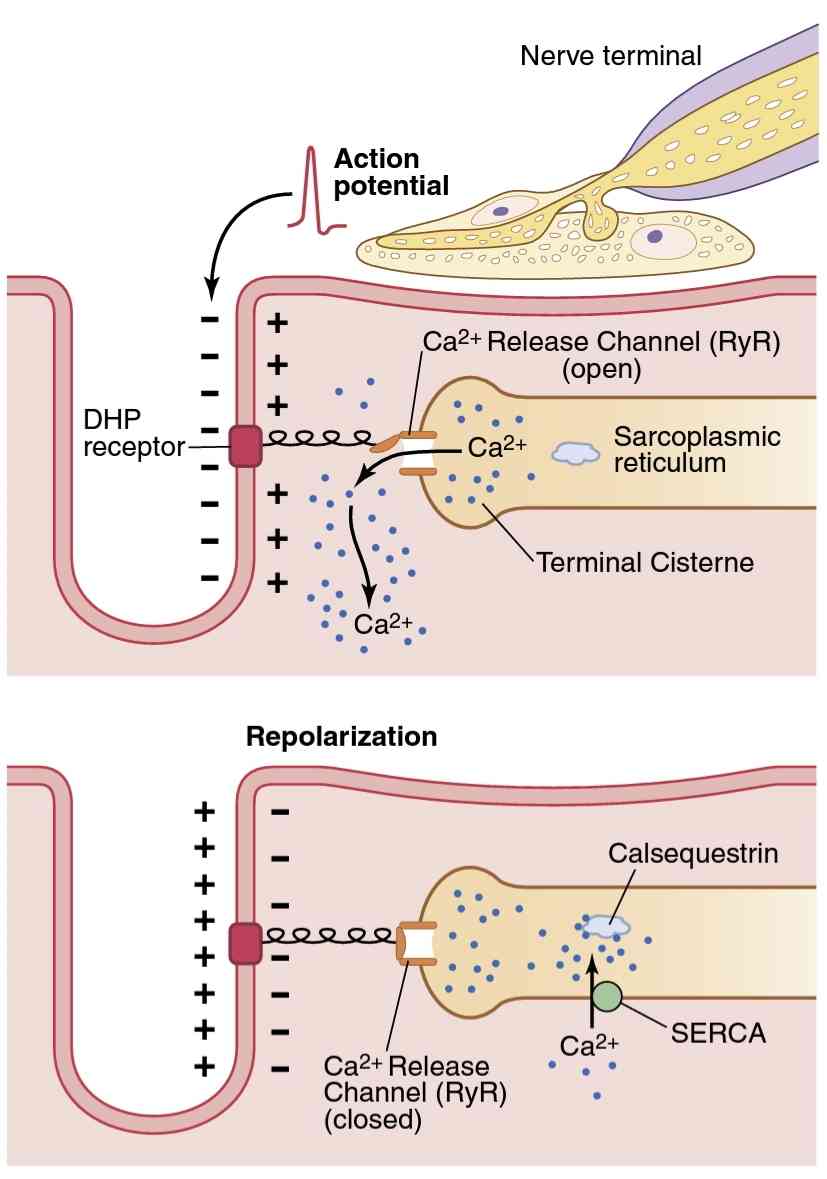
Figure 7-6.
Excitation-contraction coupling in skeletal muscle. The top panel shows an action potential in the transverse tubule that causes a conformational change in the voltage-sensing dihydropyridine (DHP) receptors, opening the ryanodine (RyR) Ca2+ release channels in the ter- minal cisternae of the sarcoplasmic reticulum and permitting Ca2+ to diffuse rapidly into the sarcoplasm and initiate muscle contraction. During repolarization (bottom panel), the conformational change in the DHP receptor closes the Ca2+ release channels, and Ca2+ is transported from the sarcoplasm into the sarcoplasmic reticulum by an adenosine triphosphate-dependent calcium pump, called SERCA (sarcoplasmic reticulum Ca2+-ATPase).
شکل 7-6.
جفت شدن تحریک – انقباض در عضله اسکلتی. پانل بالایی یک پتانسیل عمل را در لوله عرضی نشان میدهد که باعث تغییر ساختاری در گیرندههای دی هیدروپیریدینی (DHP) حسگر ولتاژ میشود، کانالهای انتشار رایانودین (RyR) Ca2+ را در مخزن انتهایی شبکه سارکوپلاسمیباز میکند و به Ca2+ اجازه میدهد تا با انقباض و انقباض Ca2+ در عضله راکوپلاسمیک منتشر شود. در طی رپلاریزاسیون (پانل پایین)، تغییر ساختاری در گیرنده DHP، کانالهای انتشار Ca2+ را میبندد و Ca2+ از سارکوپلاسم به شبکه سارکوپلاسمیتوسط یک پمپ کلسیم وابسته به آدنوزین تری فسفات به نام SERCA (شبکه سارکوپلاسمیCa2+-ATPase) منتقل میشود.
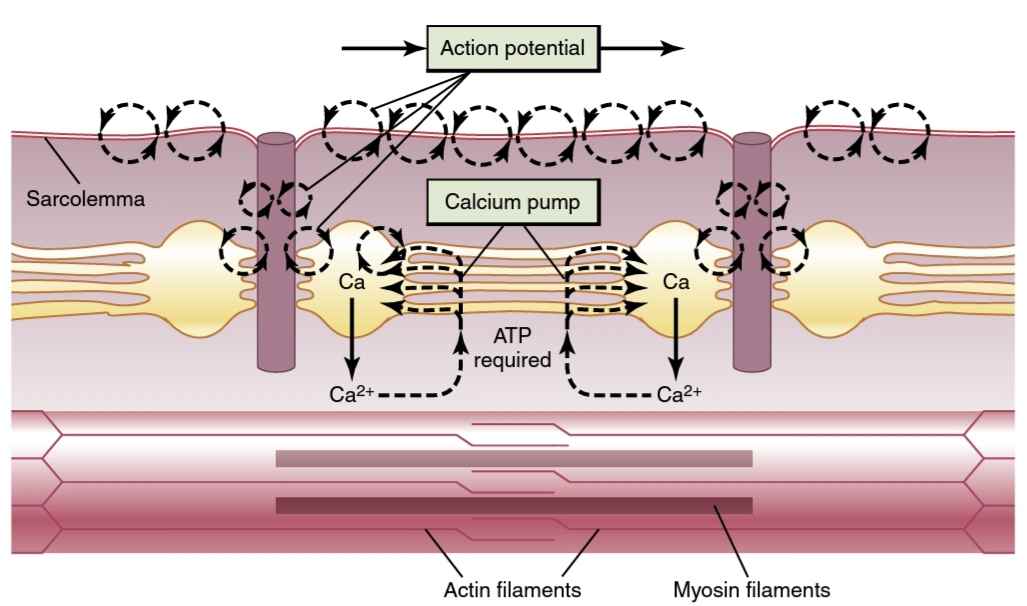
Figure 7-7.
Excitation-contraction coupling in the muscle, showing (1) an action potential that causes release of calcium ions from the sarco- plasmic reticulum and then (2) re-uptake of the calcium ions by a calcium pump. ATP, Adenosine triphosphate.
شکل 7-7.
جفت شدن تحریک-انقباض در عضله، نشان دهنده (1) پتانسیل عمل است که باعث آزاد شدن یونهای کلسیم از شبکه سارکوپلاسمیو سپس (2) جذب مجدد یونهای کلسیم توسط پمپ کلسیم میشود. ATP، آدنوزین تری فسفات.
Calcium Pump Removes Calcium lons from the Myofibrillar Fluid After Contraction Occurs. Once the calcium ions have been released from the sarcoplasmic tubules and have diffused among the myofibrils, muscle contraction continues as long as the calcium ion concen- tration remains high. However, a continually active calcium pump located in the walls of the sarcoplasmic reticulum pumps calcium ions away from the myofibrils back into the sarcoplasmic tubules (see Figure 7-6). This pump, called SERCA (sarcoplasmic reticulum Ca2+-ATPase), can concentrate the calcium ions about 10,000-fold inside the tubules. In addition, inside the reticulum is a calcium- binding protein called calsequestrin, which can bind up to 40 calcium ions for each molecule of calsequestrin.
پمپ کلسیم lons کلسیم را از مایع میوفیبریلار پس از وقوع انقباض خارج میکند. هنگامیکه یونهای کلسیم از لولههای سارکوپلاسمیآزاد شدند و در میان میوفیبریلها منتشر شدند، انقباض عضلانی تا زمانی که غلظت یون کلسیم بالا باقی بماند ادامه مییابد. با این حال، یک پمپ کلسیم به طور مداوم فعال که در دیوارههای شبکه سارکوپلاسمیقرار دارد، یونهای کلسیم را از میوفیبریلها دور میکند و به لولههای سارکوپلاسمیبرمیگرداند (شکل 7-6 را ببینید). این پمپ که SERCA (شبکه سارکوپلاسمیCa2+-ATPase) نام دارد، میتواند یونهای کلسیم را حدود 10000 برابر در داخل لولهها متمرکز کند. علاوه بر این، در داخل شبکه یک پروتئین متصل به کلسیم به نام calsequestrin وجود دارد که میتواند تا 40 یون کلسیم را برای هر مولکول calsequestrin متصل کند.
Excitatory Pulse of Calcium lons. The normal resting state concentration (<10-7 molar) of calcium ions in the cytosol that bathes the myofibrils is too little to elicit con- traction. Therefore, the troponin-tropomyosin complex keeps the actin filaments inhibited and maintains a re- laxed state of the muscle.
نبض تحریکی لونز کلسیم. غلظت طبیعی حالت استراحت (<10-7 مولار) یونهای کلسیم در سیتوزولی که میوفیبریلها را حمام میکند برای ایجاد انقباض بسیار کم است. بنابراین، کمپلکس تروپونین-تروپومیوزین رشتههای اکتین را مهار میکند و حالت آرامش عضله را حفظ میکند.
Conversely, full excitation of the T tubule and sar- coplasmic reticulum system causes enough release of calcium ions to increase the concentration in the myo- fibrillar fluid to as high as 2 × 10-4 molar concentration, a 500-fold increase, which is about 10 times the level required to cause maximum muscle contraction. Imme- diately thereafter, the calcium pump depletes the calcium ions again. The total duration of this calcium pulse in the usual skeletal muscle fiber lasts about 1/20 of a second, although it may last several times as long in some fibers and several times less in others. In heart muscle, the cal- cium pulse lasts about one-third of a second because of the long duration of the cardiac action potential.
برعکس، تحریک کامل لوله T و سیستم شبکه سارکوپلاسمیباعث آزاد شدن کافی یونهای کلسیم برای افزایش غلظت در مایع میوفیبریلار تا غلظت مولی 2×10-4 میشود که این افزایش 500 برابری است که حدود 10 برابر سطح مورد نیاز برای ایجاد حداکثر انقباض عضلانی است. بلافاصله پس از آن، پمپ کلسیم دوباره یونهای کلسیم را تخلیه میکند. مدت زمان کل این پالس کلسیم در فیبر عضلانی اسکلتی معمول حدود 1/20 ثانیه طول میکشد، اگرچه ممکن است در برخی از فیبرها چندین برابر و در برخی دیگر چندین برابر کمتر طول بکشد. در عضله قلب، نبض کلسیم در حدود یک سوم ثانیه طول میکشد زیرا مدت زمان طولانی پتانسیل عمل قلبی است.
During this calcium pulse, muscle contraction occurs. If the contraction is to continue without interruption for long intervals, a series of calcium pulses must be initiated by a continuous series of repetitive action potentials, as discussed in Chapter 6.
در طول این نبض کلسیم، انقباض عضلانی رخ میدهد. اگر قرار است انقباض بدون وقفه برای فواصل طولانی ادامه یابد، یک سری پالسهای کلسیم باید با یک سری پتانسیلهای تکراری عمل مداوم آغاز شود، همانطور که در فصل 6 بحث شد.
Malignant Hyperthermia
In susceptible individuals, malignant hyperthermia and a hypermetabolic crisis may be triggered by exposure to certain types of anesthetics, including halothane and iso- flurane, or succinylcholine. At least six genetic mutations, especially of the ryanodine receptor or dihydropyridine receptor genes, have been shown to increase susceptibil- ity greatly to developing malignant hyperthermia during anesthesia. Little is known about the specific mechanisms whereby anesthetics interact with these abnormal recep- tors to trigger malignant hyperthermia. It is known, how- ever, that these mutations cause unregulated passage of cal- cium from the sarcoplasmic reticulum into the intracellular spaces, which in turn causes the muscle fibers to contract excessively. These sustained muscled contractions greatly increase metabolic rate, generating large amounts of heat and causing cellular acidosis, as a well as depletion of en- ergy stores.
هایپرترمیبدخیم
در افراد مستعد، هیپرترمیبدخیم و بحرانهایپرمتابولیک ممکن است با قرار گرفتن در معرض انواع خاصی از داروهای بیهوشی، از جملههالوتان و ایزوفلوران، یا سوکسینیل کولین ایجاد شود. نشان داده شده است که حداقل شش جهش ژنتیکی، به ویژه ژنهای گیرنده ریانودین یا گیرنده دی هیدروپیریدین، حساسیت به ایجاد هیپرترمیبدخیم در طول بیهوشی را افزایش میدهد. اطلاعات کمیدر مورد مکانیسمهای خاصی وجود دارد که به موجب آن بیهوشکنندهها با این گیرندههای غیرطبیعی تعامل میکنند و باعث ایجاد هیپرترمی بدخیم میشوند. با این حال، مشخص است که این جهشها باعث عبور غیرقابل تنظیم کلسیم از شبکه سارکوپلاسمیبه فضاهای درون سلولی میشوند که به نوبه خود باعث انقباض بیش از حد فیبرهای عضلانی میشود. این انقباضات عضلانی پایدار به میزان زیادی سرعت متابولیسم را افزایش میدهد، مقدار زیادی گرما ایجاد میکند و باعث اسیدوز سلولی و همچنین تخلیه ذخایر انرژی میشود.
Symptoms of malignant include muscle rigidity, high fever, and rapid heart rate. Additional complications in se- vere cases may include rapid breakdown of skeletal muscle (rhabdomyolysis) and a high plasma potassium level due to release of large amounts of potassium from damaged muscle cells. Treatment of malignant hyperthermia gener- ally involves rapid cooling and the administration of dant- rolene, a drug that antagonizes ryanodine receptors, which inhibits calcium ion release for the sarcoplasmic reticulum and thereby attenuating muscle contraction.
علائم بدخیم عبارتند از سفتی عضلانی، تب بالا و ضربان قلب سریع. عوارض اضافی در موارد شدید ممکن است شامل تجزیه سریع ماهیچههای اسکلتی (رابدومیولیز) و سطح بالای پتاسیم پلاسما به دلیل آزاد شدن مقادیر زیادی پتاسیم از سلولهای عضلانی آسیب دیده باشد. درمان هیپرترمیبدخیم معمولاً شامل خنکسازی سریع و تجویز دانترولن است، دارویی که گیرندههای رایانودین را متضاد میکند، که آزاد شدن یون کلسیم را برای شبکه سارکوپلاسمیمهار میکند و در نتیجه انقباض عضلانی را کاهش میدهد.
کلیک کنید «Bibliography: فهرست کتب مربوطه»
Also see the bibliography for Chapters 5 and 6.
Bouzat C, Sine SM. Nicotinic acetylcholine receptors at the singlechannel level. Br J Pharmacol 175:1789-1804, 2018.
Cheng H, Lederer WJ: Calcium sparks. Physiol Rev 88:1491, 2008.
Dalakas MC. Immunotherapy in myasthenia gravis in the era of biologics. Nat Rev Neurol 15:113-124, 2019.
Gilhus NE. Myasthenia gravis. N Engl J Med 37:2570-2581, 2016.
Jungbluth H, Treves S, Zorzato F, Sarkozy A, Ochala J, Sewry C, et al. Congenital myopathies: disorders of excitation-contraction coupling and muscle contraction. Nat Rev Neurol 14:151-167, 2018 Meissner G.
The structural basis of ryanodine receptor ion channel function. J Gen Physiol 149:1065-1089, 2017.
Periasamy M, Maurya SK, Sahoo SK, Singh S, Sahoo SK, Reis FCG, et al. Role of SERCA pump in muscle thermogenesis and metabolism. Compr Physiol 7:879-890, 2017.
Rekling JC, Funk GD, Bayliss DA, et al: Synaptic control of motoneuronal excitability. Physiol Rev 80:767, 2000.
Rosenberg PB: Calcium entry in skeletal muscle. J Physiol 587:3149, 2009.
Ruff RL, Lisak RP. Nature and action of antibodies in myasthenia gravis. Neurol Clin 36:275-291, 2018.
Ruff RL: Endplate contributions to the safety factor for neuromuscular transmission. Muscle Nerve 44:854, 2011.
Sine SM: End-plate acetylcholine receptor: structure, mechanism, pharmacology, and disease. Physiol Rev 92:1189, 2012.
Tintignac LA, Brenner HR, Rüegg MA. Mechanisms regulating neuromuscular junction development and function and causes of muscle wasting. Physiol Rev 95:809-852, 2015
Vincent A: Unraveling the pathogenesis of myasthenia gravis. Nat Rev Immunol 10:797, 2002.
🚀 با ما همراه شوید!
تازهترین مطالب و آموزشهای مغز و اعصاب را از دست ندهید. با فالو کردن کانال تلگرام آیندهنگاران مغز، از ما حمایت کنید!

 ورود/ ثبت نام با جیمیل
ورود/ ثبت نام با جیمیل
